 |
|
Bios
Dr. Light a clinical vitreo- Dr. Hsu is with Mid DISCLOSURES: Drs. Light and Hsu have no relevant financial relationships to disclose. |
A 68-year-old man with history of hypertension, type 2 diabetes (without prior retinopathy) and psoriasis presented to the emergency department for blunt trauma to the right eye following an assault. His ocular history was negative aside from a history of uncomplicated cataract extraction with intraocular lens placement in both eyes eight years earlier.
Ophthalmic consultation revealed findings consistent with traumatic mydriasis and iritis in the affected right eye. Topical prednisolone and cycloplegics were prescribed. However, a dilated exam revealed abnormalities of both fundi, so the patient was referred to the ophthalmology clinic for further evaluation.
 |
Clinical evaluation
On evaluation in the clinic one week later, visual acuity with correction was 20/40 in both eyes. Intraocular pressures were 17 mmHg OD and 22 mmHg OS. The right pupil was dilated and sluggish, consistent with prior diagnosis of traumatic mydriasis. There was no relative afferent pupillary defect by reverse testing.
An anterior segment examination of both eyes revealed well-centered posterior chamber intraocular lenses with trace pigment in the formed anterior vitreous, but no inflammatory cells or vitritis.
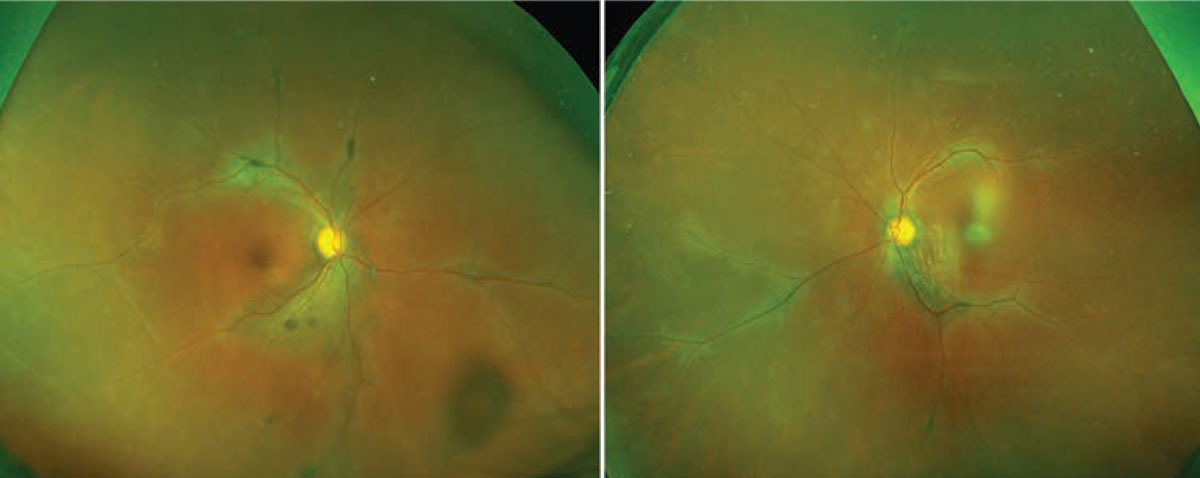
The fundus examination showed bilateral hypopigmentation emanating from the discs and extending out along the major superior and inferior arcades, with additional hypopigmentation noted along the nasal venules (Figure 1). Clumps of hyperpigmentation were also seen within the regions of hypopigmentation and were concentrated adjacent to the major retinal veins. A full scleral-depression exam showed no retinal tears or detachments or any vitreous snowballs or snow-banking in either eye.
 |
|
Figure 1. Fundus examination shows multifocal areas of subretinal fluid bilaterally. |
Role of multimodal imaging
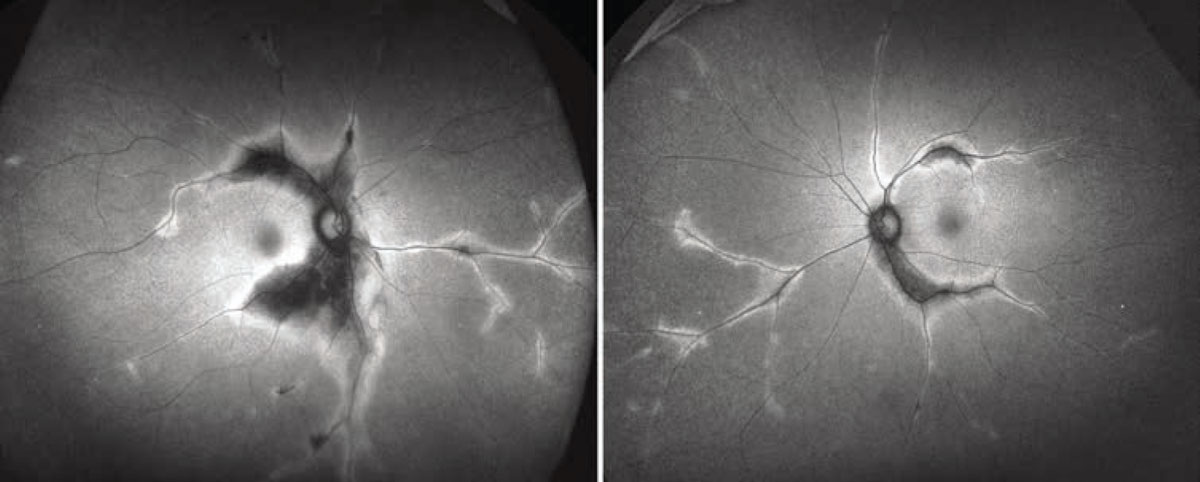
Multimodal imaging helped to further characterize the pigmentary changes. Widefield fundus autofluorescence imaging was notable for marked hypoautofluorescence corresponding to the regions of hypopigmentation described on the fundus exam, with strongly hyperautofluorescent borders (Figure 2)
 |
|
Figure 2. Widefield fundus autofluorescent imaging of the right and left fundi demonstrate marked hypoautofluorescence that corresponds to the hypopigmentation seen on color fundus exam. The lesions show well-defined hyperautofluorescent borders. |
Optical coherence tomography scans in the central macula showed preservation of normal inner and outer retinal lamination patterns (Figures 3A,B). But severe atrophy of the retinal pigment epithelium and choriocapillaris, with associated photoreceptor layer loss, were evident in the areas of peripheral macular pigmentary change (Figures 3C,D). No subretinal or intraretinal fluid was apparent.
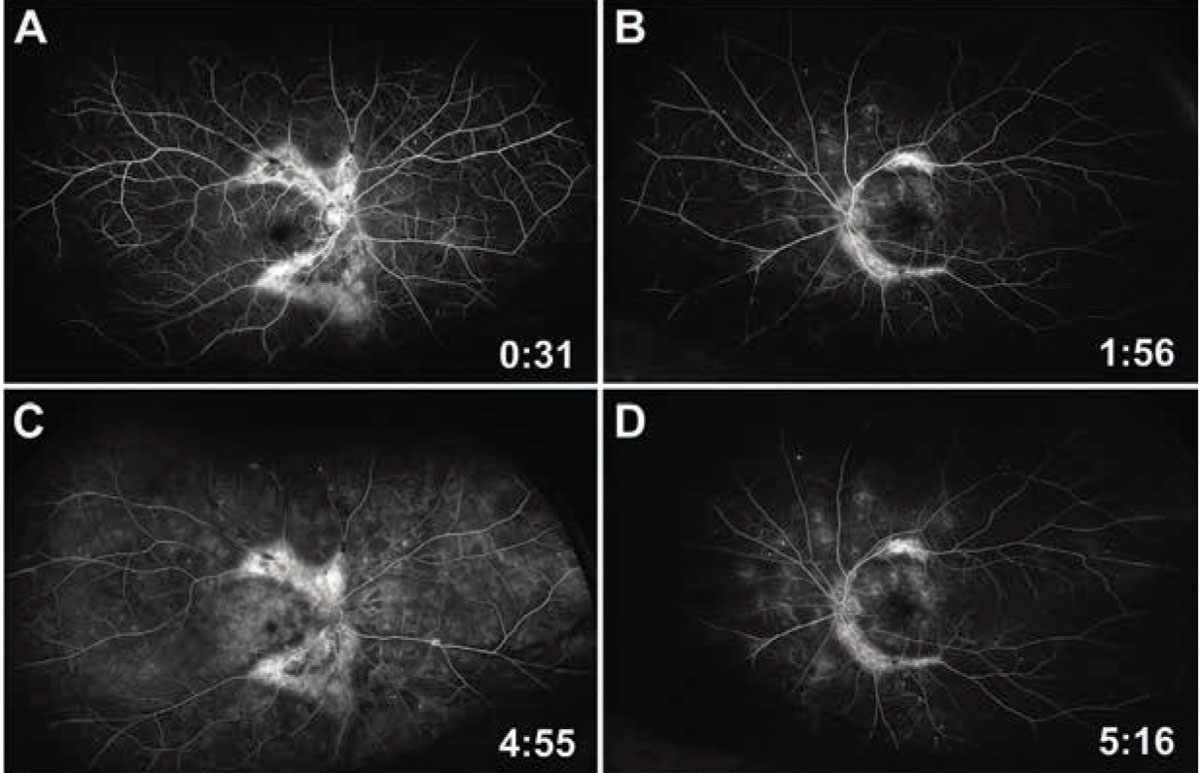
We also obtained widefield fluorescein angiography. Early frames revealed normal arterial and venous filling times with scattered microaneurysms and areas of peripheral capillary dropout, consistent with subclinical nonproliferative diabetic retinopathy. Large areas of hyperfluorescence, consistent with window defects, were seen along the proximal arcades (Figures 4A,B). Late frames demonstrated patchy interstitial leakage in the macula and periphery with persistent window defects and/or staining in the regions of fundus pigmentary changes (Figures 4C,D). Hyperpigmented clumps corresponded to signal blockage on the angiogram. No disc or vascular leakage was apparent in either eye.
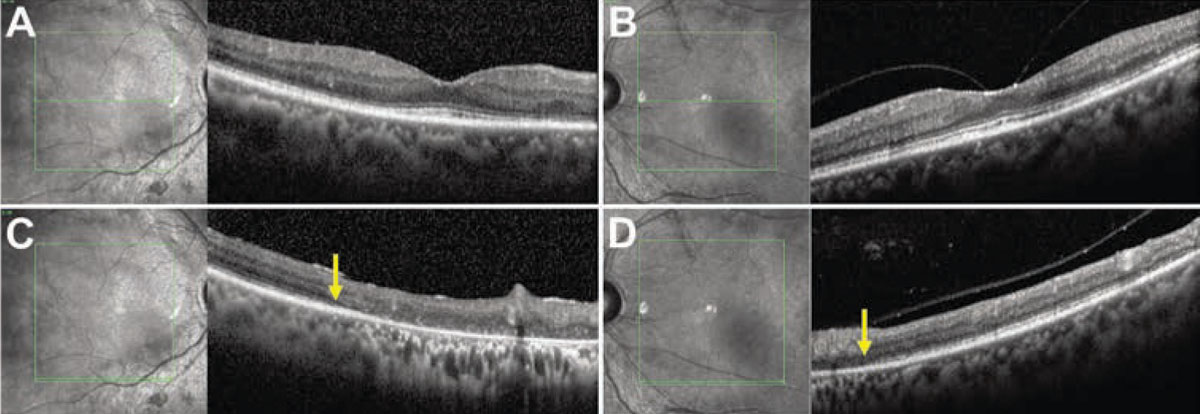 |
| Figure 3. Spectral-domain optical coherence tomography imaging of the right and left maculae with corresponding en face infrared reflectance images show preservation of outer retinal lamination patterns in the central macula and fovea in the right (A) and left (B) eyes, respectively. Line scans through the right (C) and left (D) inferior maculae show outer-retinal atrophy nasally (arrows indicate border of intact and missing external limiting membrane), corresponding to the hypopigmentation seen on the fundus exam. |
Due to the borderline cup-to-disc ratios and asymmetric tonometry mentioned previously, we obtained a 24-2 Humphrey visual field. While no glaucomatous defects were seen, the results demonstrated profound sensitivity loss in a temporal horseshoe-like pattern, corresponding topographically to the areas of outer retinal atrophy (Figure 5).
 |
|
Figure 4. Early-to-mid frames of widefield fluorescein angiography of the right (A) and left (B) eye show clear window defects in the areas of fundal hypo-pigmentation, with intact arterial and venous filling. Late frames (C and D) reveal persistent window defects and staining. Background nonproliferative diabetic retinopathy and subclinical macular edema are also seen in both eyes. |
Broad differential, classic look
Given the large areas of outer retinal, RPE and choriocapillaris atrophy along with significant visual field loss, we carefully considered a differential diagnosis that included infectious, inflammatory, dystrophic and degenerative etiologies.
The atrophic changes concentrated in the vascular distribution suggested the possibility of a “burned out” infectious or inflammatory vasculitis, which may be associated with herpes viruses, syphilis, tuberculosis, sarcoidosis or Behçet’s disease. We also considered serpiginous choroiditis and acute zonal occult outer retinopathy (AZOOR). In the category of dystrophies or degenerations, a retinitis pigmentosa variant, cone-rod dystrophies and atypical geographic atrophy were candidate diagnoses.
Ultimately, after a literature review and consultation with uveitis specialists, we concluded the findings were classic for pigmented paravenous retinochoroidal atrophy (PPRCA).
Pathology of PPRCA
PPRCA was first described in 1937 by T. Hewiston Brown, MC, ChB, in a 47-year-old man with alopecia areata.1 Since its initial description, this clinical entity has been described by several different names, including retinochoroiditis radiata, pseudoretinitis pigmentosa, chorioretinitis striata, and congenital pigmentation/melanosis of the retina.2
Clinical features are fairly stereotypical and, as seen in our patient, include pigment clumping along retinal venules with associated retinochoroidal atrophy, often involving the peripapillary region, with macular sparing. The lesions are considered to be nonprogressive or only slowly progressive.3
Interestingly, the majority of case reports of PPRCA have been in male patients,4 though the reason for a sex-specific predilection has not been elucidated.
The definitive location of the instigating pathology has not been established, though it has been hypothesized that the disease process has its origin in the RPE.5 Hypoperfusion and loss of the choriocapillaris layer have been described within areas of pigmentary atrophy in the form of indocyanine green angiography hypocyanescence and OCT angiography flow voids. However, it’s unclear whether this phenomenon triggers the outer retinal degeneration or is secondary to it.6
What’s more, while both FA and ICG-A demonstrate clear RPE and choriocapillaris atrophy, ICG-A has been shown to better detect regions of disease involvement that aren’t yet apparent on FA imaging.7
Etiology of PPRCA
As its name implies, PPRCA is a descriptive term that characterizes the relevant clinical exam and imaging findings but doesn’t give insight into its etiologic underpinnings. Early case reports proposed associations with infectious processes, including tuberculosis,1 congenital syphilis8 and measles.5,9 Others suggested a primary dysgenesis or degenerative process.10-12
An interesting published longitudinal case described a 17-year-old girl who developed bilateral sequential acute vision loss with marked retinal edema of the posterior pole.4 Over the course of the next 20 years, she developed retinal pigmentary changes typical of PPRCA, suggesting that a remote inflammatory insult may indeed be an inciting factor, resulting in this unique clinical phenotype.
However, a comprehensive review proposed that while the findings in PPRCA may be the common end result of any number of infectious or inflammatory insults, the term should be reserved for primary, idiopathic cases, while those cases secondary to other pathology should be designated pseudo-PPRCA.2
Investigations in the past few decades have also raised the possibility of an underlying genetic mechanism for PPRCA. Gareth McKay, PhD, and colleagues reported a series of autosomal dominantly inherited cases of PPRCA phenotype in a family with a confirmed mutation in the CRB1 gene, albeit with variable expression among family members and higher severity in the males.13
Other reports have described additional clusters of familial cases, with speculated inheritance ranging from autosomal-dominant or recessive, to X- and even Y-linked patterns.2 Nevertheless, discordant expression of PPRCA between monozygotic twins has been reported, confirming the complex nature of this clinical phenotype.14
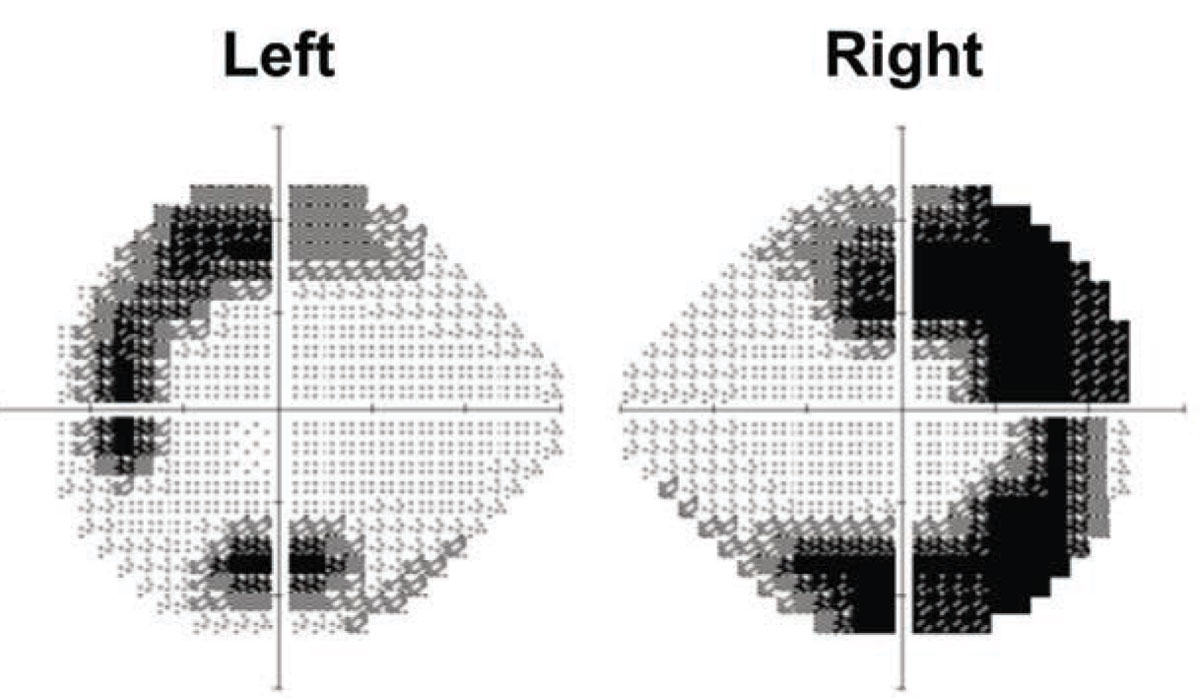 |
| Figure 5. Humphrey visual fields of both eyes with 24-2 protocol. Arcuate temporal field defects crossing both the horizontal and vertical midlines demonstrate greater severity in the right eye than the left. |
Follow-up
Our patient was re-examined at increasing intervals over the next year and found to have complete stability of the pigmentary changes and no decrease in visual acuity.
Cognizant of our inability to distinguish primary, idiopathic PPRCA from
pseudo-PPRCA based on clinical phenotype alone, we ordered a focused work-up including tuberculosis and syphilis serologies, as well as chest radiography to rule-out sarcoid-associated hilar lymphadenopathy. Testing revealed no underlying systemic conditions. Following prompt resolution of his initial traumatic iritis, the patient demonstrated no evidence of ocular inflammation at any point during follow-up.
Bottom line
PPRCA is a rare disease, often detected incidentally on routine or unrelated fundus examination. At worst, it’s slowly progressive, rarely involving the macula and central vision. At best, it’s stationary and asymptomatic for the patient. Multimodal imaging is very useful for confirming its characteristic appearance.
Given the lack of clear etiologic underpinnings and the distinct likelihood that multiple factors, including those of an infectious or inflammatory nature, may result in a PPRCA-like phenotype, a clinician confronted with such a presentation should obtain a thorough medical history, maintain high clinical suspicion and a broad differential diagnosis, and make efforts to rule out potentially undiagnosed and treatable ocular and systemic conditions. RS
The authors acknowledge Bryn Burkholder, MD, uveitis specialist, and Ravi Pandit, MD, retina specialist, of the Wilmer Eye Institute, Johns Hopkins University, Baltimore, for their expert consultation in this case.
REFERENCES
1. Brown TH. Retino‑choroiditis radiata. Br J Ophthalmol. 1937;21: 645‑648.
2. Huang HB, Zhang YX. Pigmented paravenous retinochoroidal atrophy (Review). Exp Ther Med. 2014;7:1439-1445.
3. Choi JY, Sandberg MA, Berson EL. Natural course of ocular function in pigmented paravenous retinochoroidal atrophy. Am J Ophthalmol. 2006;141:763-765.
4. Murray AT, Kirkby GR. Pigmented paravenous retinochoroidal atrophy: A literature review supported by a unique case and insight. Eye (Lond). 2000;14 Pt 5:711-716.
5. Chisholm IA, Dudgeon J. Pigmented paravenous retino-choroidal atrophy. Helicoid retino-choriodal atrophy. Br J Ophthalmol. 1973;57:584-587.
6. Shen Y, Xu X, Cao H. Pigmented paravenous retinochoroidal atrophy: A case report. BMC Ophthalmol. 2018;18:136.
7. Yanagi Y, Okajima O, Mori M. Indocyanine green angio-graphy in pigmented paravenous retinochoroidal atrophy. Acta Ophthalmol Scand. 2003;81:60-67.
8. Hsin-Hsiang C. Retinochoroiditis radiata. Am J Ophthalmol. 1948;31:1485-1487.
9. Foxman SG, Heckenlively JR, Sinclair SH. Rubeola retinopathy and pigmented paravenous retinochoroidal atrophy. Am J Ophthalmol. 1985;99:605-606.
10. Weve H. Degeneration retinae paravenosa. Bibl Ophthalmol. 1957;12:664-670.
11. Brognoli C. Due casi di atrofia coroideale progressiva [Two cases of progressive choroidal atrophy]. Ann Ottalmol Clin Ocul. 1952;78:259-68.
12. Franceschetti A. A curious affection of the fundus oculi: Helicoid peripapillar chorioretinal degeneration. Its relation to pigmentary paravenous chorioretinal degeneration. Doc Ophthalmol. 1962;16:81-110.
13. McKay GJ, Clarke S, Davis JA, Simpson DA, Silvestri G. Pigmented paravenous chorioretinal atrophy is associated with a mutation within the crumbs homolog 1 (CRB1) gene. Invest Ophthalmol Vis Sci. 2005;46:322-328.
14. Small KW, Anderson WB Jr. Pigmented paravenous retinochoroidal atrophy. Discordant expression in mono-zygotic twins. Arch Ophthalmol. 1991;109:1408-1410.



