 |
History
The patient recalled an episode of anterior uveitis in both eyes several years before. A work-up at the time was negative. He was taking difluprednate b.i.d. OU as prophylaxis and denied recurrence of uveitis symptoms since the initial episode. His ocular, medical and surgical histories were otherwise negative. He had no allergies and was not taking any medications aside from the difluprednate. The patient was a student with upcoming examinations.Examination
Best-corrected visual acuity (BCVA) was 20/20 OD and 20/200 OS. Intraocular pressure was 16 mm Hg OD and 14 mm Hg OS. Pupils were round and reactive without a relative afferent pupillary defect. Color plates were 10/10 OD and 7/10 OS. Slit lamp examination showed more posterior synechiae in the left eye, but no cell, flare or keratic precipitates. The dilated fundus exam showed scattered, faint, white, placoid lesions of less than one disc diameter along the superior arcade and temporal macula (Figure 1A). These lesions were bilateral, but more prominent and numerous OS, involving the central macula and temporal periphery (Figure 1B).Diagnosis, Workup, Treatment
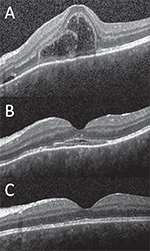
Given the characteristic white placoid lesions in both eyes, the differential diagnosis included primary inflammatory choriocapillaropathies, also referred to as the “white dot syndromes,” such as acute posterior multifocal placoid pigment epithliopathy (APMPPE) and multiple evanescent white dot syndrome (MEWDS). We also considered autoimmune etiologies such as Vogt-Koyanagi-Harada (VKH) disease, systemic lupus erythematosus (SLE) and sarcoidosis, and infectious etiologies such as tuberculosis, syphilis and Lyme disease. Optical coherence tomography (OCT) OS showed dome-shaped elevations of the ellipsoid zone (EZ) band with pockets of hyper-reflective material and subretinal fluid OS (Figure 2A, page 16). OCT OD was unremarkable.Fluorescein angiogram (FA) OS showed early hypofluorescence (Figure 1D) with late hyperfluorescence at the sites of the placoid lesions. Late frames OD showed hyperfluorescence in more regions of the temporal macula and periphery than the fundus exam (Figure 1C). Fundus autofluorescence (FAF) was largely unremarkable. Indocyanine green angiography (ICGA) showed numerous placoid foci of hypofluorescence in the temporal macula OD (Figure 1E). This finding was more prominent OS with large confluent patches of hypofluorescence covering the macula (Figure 1F). In both eyes, the number of hypofluorescent lesions was much greater than the number of lesions seen on the fundus exam and FA.
Our initial workup, which included chest x-ray and tuberculosis (PPD), syphilis, antinuclear antibody and Lyme/Bartonella/herpes simplex/cytomegalovirus titers, came back negative two days later.
We started the patient on prednisone 40 mg PO daily. One week later, best corrected visual acuity was 20/60 OS and the photopsias had resolved. We saw the patient multiple times over the next few weeks. His vision continued to improve, reaching 20/30 after one month of therapy, at which point we slowly taperedthe prednisone.
 |
Discussion
Primary inflammatory choriocapillaropathy or choriocapillaritis are terms that describe most of the diseases classified as “white dot syndromes.” They include APMPPE, MEWDS, multifocal choroiditis and serpiginous choroiditis. The disease mechanism that unifies these conditions is reduced perfusion within the choriocapillaris, presumably from localized inflammation, although the underlying etiology is not well understood. ICGA is an excellent technique to investigate suspected cases of choriocapillaritis;1 it can reveal areas of hypo- or non-perfusion within the choriocapillaris that lead to retinal ischemia and outer retinal pathology. ICGA also corresponds better to patients’ performance on functional tests such as visual fields than a fundus exam or FA.APMPPE typically presents in young adults with bilateral vision loss and may be preceded by a viral illness.2 Presenting symptoms may include photopsias, decreased vision, paracentral scotoma and metamorphopsia. Interestingly, this young patient only had unilateral symptoms despite the presence of bilateral pathology, and he did not recall any prior viral illness. Our patient displayed the classic fundus lesions that characterize APMPPE—scattered, flat, multifocal, creamy white or yellow lesions at the level of the retinal pigment epithelium (RPE). Lesions are typically located in the macula but may also involve the peripheral retina.
On FA, active lesions demonstrate characteristic early hypofluorescence and late hyperfluorescence. Early hypofluorescence is thought to result from blockage secondary to inflammation and edema of the RPE. Late hyperfluorescence results from staining of the damaged RPE and leakage from the underlying choriocapillaris. Chronic lesions show hyperfluorescence corresponding to window defects from RPE atrophy.
Our patient’s FA was consistent with the findings described here, supporting a diagnosis of APMPPE. FAF may show hypoautofluorescence of lesions in the acute phase that persists even with lesion resolution. Hyperautofluorescence may also occur late due to deposition of lipofuscin or altered metabolism of affected RPE.5 This particular case of APMPPE did not demonstrate such changes on FAF.
Recent analyses of APMPPE lesions using spectral-domain OCT have demonstrated various stages of retinal changes.3 At onset, placoid lesions appear as prominent, dome-shaped elevations of the EZ band, with hyper-reflective material and subretinal fluid accumulation, as OCT of our patient’s left eye demonstrated.4 Over the disease course, the dome-shaped lesion flattens, the EZ band thickens, the outer nuclear layer shows hyper-reflectivity followed by thinning and the RPE thickens.5 After a few months, restoration of the outer retina occurs, as does reconstitution of the EZ band and the RPE sustains minimal residual irregularity. Our patient’s OCT OS appeared to follow this course, with improvement of vision corresponding to decreased subretinal fluid and restoration of the EZ band.
 |
| Dr. Olmos de Koo is an assistant professor of ophthalmology at University of Southern California Eye Institute and the director of the vitreoretinal fellowship at the Keck School of Medicine of USC in Los Angeles. Dr. Xu is an ophthalmology resident at USC Eye Institute/Los Angeles County + USC program. Dr. George is a vitreoretinal surgical fellow at USC Eye Institute. |
Because of our patient’s functional requirements, foveal involvement OS and extent of vision loss OS, we decided to treat early with oral prednisone. Uncommon cases of severe vision loss from persistent RPE alterations in the fovea have been reported. Choroidal neovascularization can also complicate APMPPE. Recurrences of APMPPE are rare.
Finally, several reports have linked APMPPE to central nervous system vasculitis, with manifestations ranging from headaches to venous sinus thrombosis.7 The combination of APMPPE with neurologic manifestations occurs more frequently in men and very rarely can have dire consequences, including death. Thus, a thorough review of systems is critical for any patient presenting with a possible diagnosis of APMPPE. Treatment recommendations in such cases include IV corticosteroids followed by a slow oral taper in combination with an immunosuppressant.
Conclusion
This case demonstrates the multiple classic exam and imaging findings associated with APMPPE. It also highlights the utility of ICGA in the diagnosis of APMPPE and other forms of choriocapillaritis. It underscores the importance of performing a full work-up for autoimmune and infectious etiologies before arriving at the diagnosis of APMPPE. Steroid treatment may offer a more favorable prognosis when the fovea is involved. RSReferences
1. Cimino L, Auer C, Herbort CP. Sensitivity of indocyanine green angiography for the follow-up of active inflammatory choriocapillaropathies. Ocul. Immunol. Inflamm. 2000;8:275–283.2. Gass JD. Acute posterior multifocal placoid pigment epitheliopathy. Arch. Ophthalmol. 1968;80:177–185.
3. Goldenberg D, Habot-Wilner Z, Loewenstein A, Goldstein M. Spectral domain optical coherence tomography classification of acute posterior multifocal placoid pigment epitheliopathy. Retina. 2012;32,1403–1410.
4. Scheufele TA, Witkin AJ, Schocket LS, et al. Photoreceptor atrophy in acute posterior multifocal placoid pigment epitheliopathy demonstrated by optical coherence tomography. Retina. 2005;25: 1109–1112.
5. Souka AA, Hillenkamp J, Gora F, Gabel VP, Framme C. Correlation between optical coherence tomography and autofluorescence in acute posterior multifocal placoid pigment epitheliopathy. Graefes Arch Clin Exp Ophthalmol. 2006;244: 1219–1223.
6. Fiore T, Iaccheri B, Androudi S, et al. Acute posterior multifocal placoid pigment epitheliopathy: outcome and visual prognosis. Retina. 2009;29:994–1001.
7. O’Halloran HS, Berger JR, Lee WB, et al. Acute multifocal placoid pigment epitheliopathy and central nervous system involvement: nine new cases and a review of the literature. Ophthalmology. 2001;108,861–868.



