The American Academy of Ophthalmology supports genetic testing for all patients with a presumed or suspected inherited, or Mendelian, retinal disease.1 The AAO Task Force on Genetic Testing states that clinicians should avoid routine genetic testing for genetically complex disorders such as age-related macular degeneration.
 |
The benefits of genetic testing for inherited genetic disease are myriad: A positive genetic test can elucidate or confirm a diagnosis and provide information about likely prognosis. Increasingly, genetic testing is used to screen patients as possible candidates for clinical treatment trials.
Gene-based clinical trials are in progress for some types of Usher syndrome, Leber congenital amaurosis (LCA), retinitis pigmentosa (RP), Stargardt macular dystrophy and choroideremia. Several clinical trials of gene therapy for RPE65-related disease have concluded; the Food and Drug Administration this year could approve the first gene therapy for clinical use.2,3
How Genetic Testing Can Alter Medical Management
In the absence of approved gene-based treatments, though, genetic testing can still change medical management in these meaningful ways (Diagram, below):
• Reduce the need for additional electrophysiology and/or serological testing clinicians use to narrow differential diagnoses.
• Clarify the need for medical imaging and surveillance associated with syndromic disease.
• Provide guidance in determining ocular surveillance.1
• Stratify patients based on risk factors for neovascularization or cystoid macular edema.4,5
• Determine when to change medications and supplements, such as when to direct patients with
ABCA4-macular dystrophy to avoid supplemental vitamin A.6
Testing Clarifies Inheritance
Of
 |
These discrepancies in underlying inheritance can result from several genetic factors, including de novo dominant mutations, X-linked disease in families with manifesting female carriers, dominant disease with reduced penetrance and pseudodominance.7 These complex genetic factors, often dismissed as too rare for consideration in the typical practice, collectively account for a significant percentage of the underlying genetics of inherited retinal disease.
Genetic Complexity
In 1984, Shom Shanker Bhattacharya, PhD, and colleagues mapped the RP2 gene associated with X-linked RP.8 The ensuing 30-plus years have seen tremendous advances. Today, nearly 300 inherited retinal disease genes have been mapped, and more than 250 of those genes have been cloned.9
Retinal disease genetics have proven to be a model of genetic complexity. They display not only allelic heterogeneity, where many different disease-causing mutations are found within a particular gene, but also:
• Genetic heterogeneity—that is, mutations in different genes may cause the same disease.
• Phenotypic heterogeneity—different mutations within the same gene may produce different clinical phenotypes.
• Clinical heterogeneity—the same mutation in different individuals, even within the same family, may produce different clinical consequences.
RP is
 |
For example, some children with LCA, presenting in infancy or early childhood with apparently isolated retinal disease, may actually have an underlying diagnosis of a syndromic condition such as Joubert syndrome. Prior to multi-gene testing, young children with LCA typically had an MRI to rule out brain abnormalities associated with some of the syndromic forms of LCA.10 Today, clinicians typically use genetic testing to guide surveillance. Given known genotype-phenotype data, some patients will benefit from MRI, renal ultrasound, hearing evaluations or diabetes monitoring; others will not.
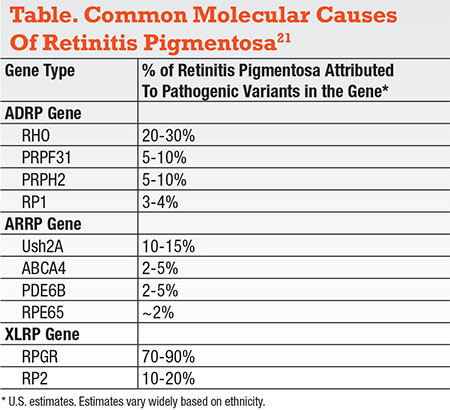
Conversely, some genes historically associated with syndromic forms of retinal dystrophy can in fact cause isolated RP or LCA (Table). This has significant implications for the interpretation of genetic testing data. While Ush2A is the gene most commonly known to be associated with Usher syndrome type 2, it is also now known to be the most common cause of nonsyndromic RP.11
The CLN3 gene has long been known to be associated with neuronal ceroid lipofuscinosis (NCL, or Batten disease), a universally fatal disease that typically begins with childhood onset of severe retinal dystrophy, followed by rapid psychomotor deterioration. Recently though, mutations in CLN3 have been described in multiple unrelated families with nonsyndromic RP.12 Genes previously believed to cause a very narrow syndromic phenotype may in fact be associated with a high degree of variability in symptom type, onset, severity and progression.13
Due to the wide range of phenotypic and clinical heterogeneity known to occur in some genetic diseases—including, but not limited to inherited retinal diseases—a significant shift is underway in medical genetics to begin naming diseases according to their underlying molecular cause. Mutations in RPE65 are known to be associated with not only LCA but also severe early childhood onset retinal dystrophy and juvenile onset RP.14,15 CRB1 mutations have been associated with LCA, RP, cone-rod dystrophy and RP with preserved para-arteriolar retinal pigment epithelium. Gene-based treatments should benefit patients with underlying mutations in those genes irrespective of their narrowly defined clinical diagnosis.
Laboratory Genetics
For more than 20 years, genetic testing was performed almost exclusively on a single-gene or targeted genetic mutation basis. To date, most patients have been tested for only a few genes that the ordering physician determines are the most statistically likely causative genes. With the advent and application of next-generation sequencing technology, and in some labs whole-exome sequencing, clinicians now routinely order testing for more than 150 genes in a single test.12,16 As a result, the likelihood of a positive test has risen dramatically. A causative mutation can be identified in 60 to 80 percent of patients with a clinical diagnosis of RP.16,17
As is frequently the case, though, more is not always better. With additional genetic testing comes greater complexity of results. Today, in nearly every patient tested for inherited retinal disease, panel testing identifies and reports multiple “variants of uncertain significance.” With time, the vast majority of these identified variants will be reclassified as normal genetic variation, but currently they often generate confusion for the patient and create a dilemma for the physician who doesn’t have the time to verify and explain those findings. For this reason (as well as cost), the recommendation
 |
What is “unnecessary parallel” testing? The 2016 Recommendations on Clinical Assessment of Patients with Inherited Retinal Degeneration state: “Multi-gene testing is typically necessary for the successful molecular diagnosis of a disease such as retinitis pigmentosa, where >100 causative genes are known.”18
In some situations, though, large-panel testing is unnecessary, at least as a first-line test. X-linked retinoschisis and Best macular dystrophy are examples of conditions where testing of a single gene is often sufficient to confirm a patient’s clinical diagnosis. For achromatopsia, for which testing of a small number of genes will identify the cause of disease in more than 80 percent of cases, a small panel typically suffices.
Importantly, physicians should utilize testing that follows standard guidelines for the interpretation and disclosure of genetic variants. In 2015, the American College of Medical Genetics and the Association for Molecular Pathology published a joint consensus document with guidelines for the interpretation of sequence variants.19 Those guidelines provide a framework for greater standardization of the interpretation and use of variants identified in genetic testing.
Who Should Order and Interpret Genetic Tests?
The AAO task force states: “Ophthalmologists who order genetic tests should either provide genetic counseling to their patients themselves, if qualified to do so, or ensure that counseling is provided by a trained individual such as a board-certified medical geneticist or genetic counselor.”
However, this recommendation has met with some controversy. Some professionals believe that only medical geneticists or ophthalmologists with board certification in medical genetics should order genetic testing for inherited retinal disease patients.20
Others disagree, stating that there are not enough board-certified medical geneticists to handle the patient volume, and retina specialists already diagnose and counsel patients with inherited retinal diseases using methods other than genetic testing, such as electrophysiology. In addition, as the availability of gene-based treatments increases, a larger number of retina specialists will want to effectively screen their own patients.
Even motivated retina specialists, though, eager to screen patients as candidates for gene-based treatments, struggle to meet the demands of incorporating genetic testing into their medical practices. Many physicians bridge this need by partnering with genetic counselors, who can collect detailed family histories, provide genetic and prognostic counseling, and review molecular testing options with patients. Genetic counselors carefully review genetic variants in test reports and research their potential contribution to disease. Following genetic testing, the genetic counselor may coordinate additional testing of family members, enroll the patient in research or patient registries and discuss clinical trials.
Patients and providers agree that genetic testing can have a multitude of benefits. Besides the clear benefits to patient management, patients and families often describe a peace of mind that comes at the end of a sometimes long diagnostic odyssey. They frequently feel more engaged in the disease community and more empowered to participate in (and comply with) their health care.
But psychosocial risks to genetic testing exist as well. Patients sometimes describe an emotional burden from learning that their family members may carry a genetic disease. Others experience intense disappointment when genetic testing does not indicate that they will qualify for a clinical trial. Younger patients may struggle with family planning decisions.
For all of these reasons, patients receiving genetic testing results deserve the time of a trained professional who can explain the findings and identify individuals in need of additional support. RS
REFERENCES
1. Recommendations on clinical assessments of patients with inherited retinal degenerations-2016. American Academy of Ophthalmology website. https://www.aao.org/clinical-statement/recommendations-on-clinical-assessment-of-patients Updated June 2016. Accessed February 3, 2017.
2. Cideciyan AV, Hauswirth WW, Aleman TS, et al. Human RPE65 gene therapy for Leber congenital amaurosis: persistence of early visual improvements and safety at 1 year. Hum Gene Ther. 2009;20:999-1004.
3. Jacobson SG, Cideciyan AV, Ratnakaram R, et al. Gene therapy for Leber congenital amaurosis caused by RPE65 mutations: safety and efficacy in 15 children and adults followed up to 3 years. Arch Ophthalmol. 2012;130:9-24.
4. Gliem M, Muller PL, Mangold E, et al. Sorsby fundus dystrophy : novel mutations, novel phenotypic characteristics, and treatment outcomes. Invest Ophthal Vis Sci. 2015;56:2664-2676.
5. Dantas MA, Slakter JS, Negrao S, Fonseca RA, Kaga T, Yannuzzi LA. Photodynamic therapy with verteporfin in mallatia leventinese. Ophthalmology. 2009;109:296-301.
6. Radu RA, Yuan Q, Hu J, et al. Accelerated accumulation of lipofuscin pigments in the RPE of a mouse model for ABCA4-mediated retinal dystrophies following Vitamin A supplementation. Invest Ophthalmol Vis Sci. 2008;49:3821-3829.
7. Branham K, Huang J, Jayasundera K, Heckenlively J. Inheritance of retinitis pigmentosa: Update in the era of genetic testing. Invest Ophthalmol Vis Sci. 2013;54:4019.
8. Bhattacharya SS, Wright AF, Clayton JF, et al. Close genetic linkage between X-linked retinitis pigmentosa and a restriction fragment length polymorphism identified by recombinant DNA probe L1.28. Nature. 1984:309;253–255.
9. RetNet: Summaries of genes and loci causing retinal disease. https://sph.uth.edu/RetNet/sum-dis.htm#D-graph. Updated January 24, 2017. Accessed February 3, 2017.
10. Traboulsi EI, Koenekoop R, Stone EM. Lumpers or splitters? The role of molecular diagnosis in Leber congenital amaurosis. Ophthalmic Genet. 2009;27:113-115.
11. Lenassi E, Vincent A, Li Z, et al. A detailed clinical and molecular survey of subjects with nonsyndromic USH2A retinopathy reveals an allelic hierarchy of disease-causing variants. Eur J Hum Genet. 2015:23;1318–1327.
12. Wang F, Wang H, Tuan HF, et al. Next generation sequencing‐based molecular diagnosis of retinitis pigmentosa: identification of a novel genotype‐phenotype correlation and clinical refinements. Hum Genet. 2014;133:331–345.
13. Warrier V, Vieira M, Mole S. Genetic basis and phenotypic correlations of the neuronal ceroid lipofusinoses. Biochim Biophysica Acta. 2013;1832:1827–1830.
14. Weleber RG, Michaelides M, Trzupek KM, et al. The phenotype of Severe Early Childhood Onset Retinal Dystrophy (SECORD) from mutation of RPE65 and differentiation from Leber congenital amaurosis. Invest Ophthalmol Vis Sci. 2011;52:292-302.
15. Thompson DA, Gyurus P, Fleischer LL, et al. Genetics and Phenotypes of RPE65 mutations in inherited retinal degeneration. Invest Ophthalmol Vis Sci. 2000;41:4293-4299.
16. Consugar MB, Navarro-Gomez D, Place EM, et al. Panel-based genetic diagnostic testing for inherited eye diseases is highly accurate and reproducible, and more sensitive for variant detection, than exome sequencing. Genet Med. 2014;17:253-261.
17. Eisenberger T, Neuhaus C, Kahn AO, et al. Increasing the yield in targeted next-generation sequencing by implicating CNV analysis, non-coding exons and the overall variant load: The example of retinal dystrophies. PLOS One. 2013;8:e78496.
18. Recommendations for genetic testing of inherited eye diseases-2014. American Academy of Ophthalmology website. https://www.aao.org/clinical-statement/recommendations-genetic-testing-of-inherited-eye-d Updated February 2014. Accessed February 3, 2017.
19. Richards S, Aziz N, Bale S, et al., for the ACMG Laboratory Quality Assurance Committee. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med. 2015; 17:405–23.
20. Bateman B, Silva E. Letter to the editor: AAO task force on Genetic Testing. Ophthalmology. 2013;120:e72-73.
21. Fahim AT, Daiger SP, Weleber RG. Nonsyndromic retinitis pigmentosa overview. In: Pagon RA, Adam MD, Ardinger, HH, et al., eds. GeneReviews [Internet]. Seattle, WA: University of Washington, Seattle. 1993-2017: https://www.ncbi.nlm.nih.gov/books/NBK1417/. Accessed February 7, 2017.



