 |
 |
A 39-year-old woman presented with a two-week history of photopsias and a subjective temporal scotoma in her right eye. She denied any eye pain. Her medical history was unremarkable. Her ocular history was notable for -3.5 D spherical myopia OU. She denied any additional symptoms on a review of systems or relevant family history.
Findings upon presentation
The patient appeared generally healthy. Snellen visual acuity with correction was 20/40 OD and 20/20 OS. Intraocular pressures were 13 mmHg OD and 16 mmHg OS. Pupillary examination demonstrated round and reactive pupils without a relative afferent pupillary defect. Although a subjective scotoma had been reported, the patient demonstrated full confrontational visual fields. Slit-lamp examination was unremarkable in both eyes without evidence of anterior chamber cell or flare.
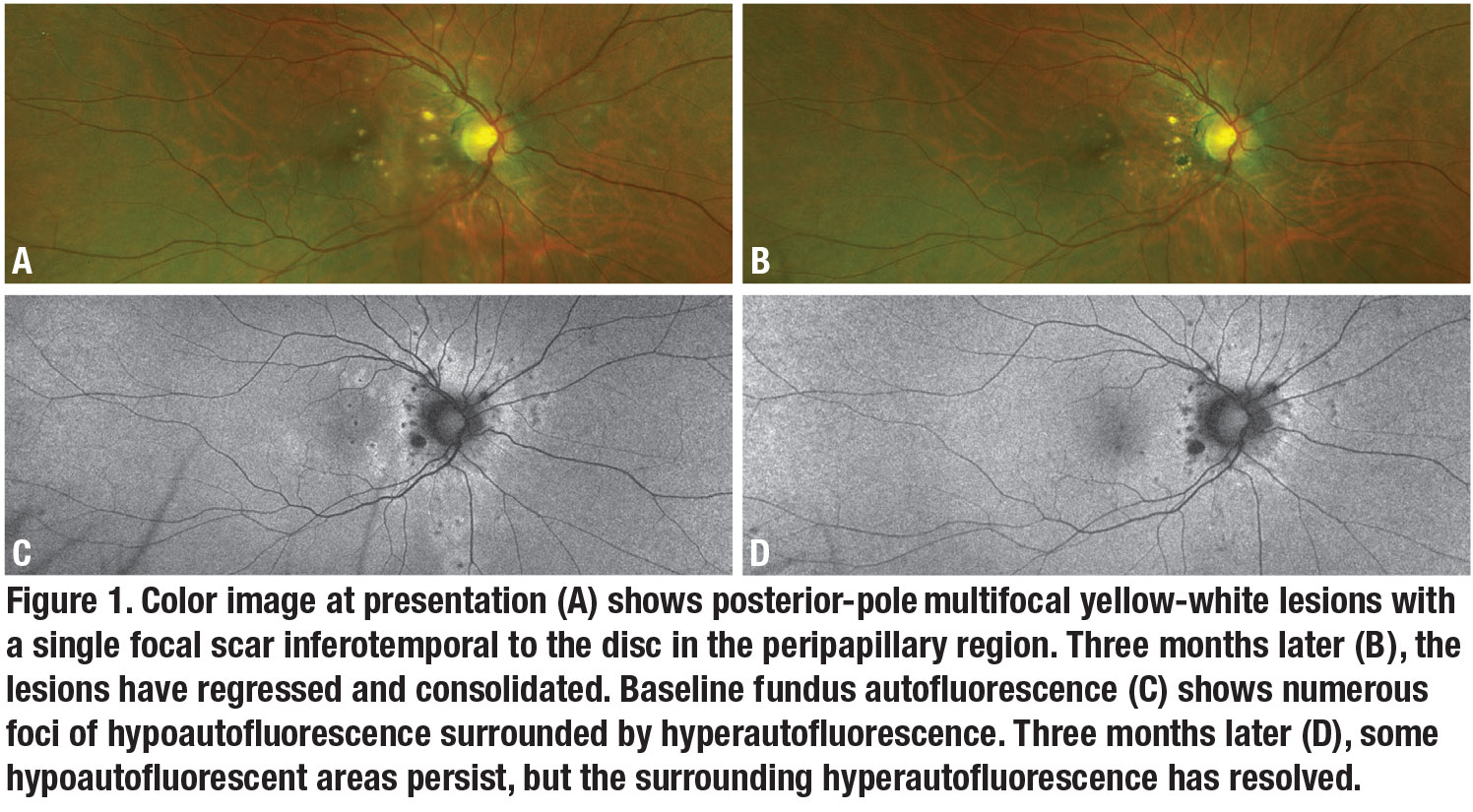
Posterior examination of the right eye revealed clear media without vitritis. We also noted punctate multifocal white-yellow retinal lesions around the optic nerve, most prominent in the nasal macula and nasal midperiphery (Figure 1A). We also saw a single area of scarring in the inferotemporal peripapillary region. Posterior examination of the left eye was unremarkable.
What imaging revealed
Fundus autofluorescence of the right eye revealed punctate hypoautofluorescence over the multifocal yellow-white lesions seen on funduscopy (Figure 1C). A ring of hyperautofluorescence surrounded the punctate areas of hypoautofluorescence, coalescing into a patchy ring of hypoautofluorescence centered around the optic disc. The temporal macula had additional faint amorphous areas of hyperautofluorescence without central hypoautofluorescence distinct from the multifocal lesions seen on funduscopy.
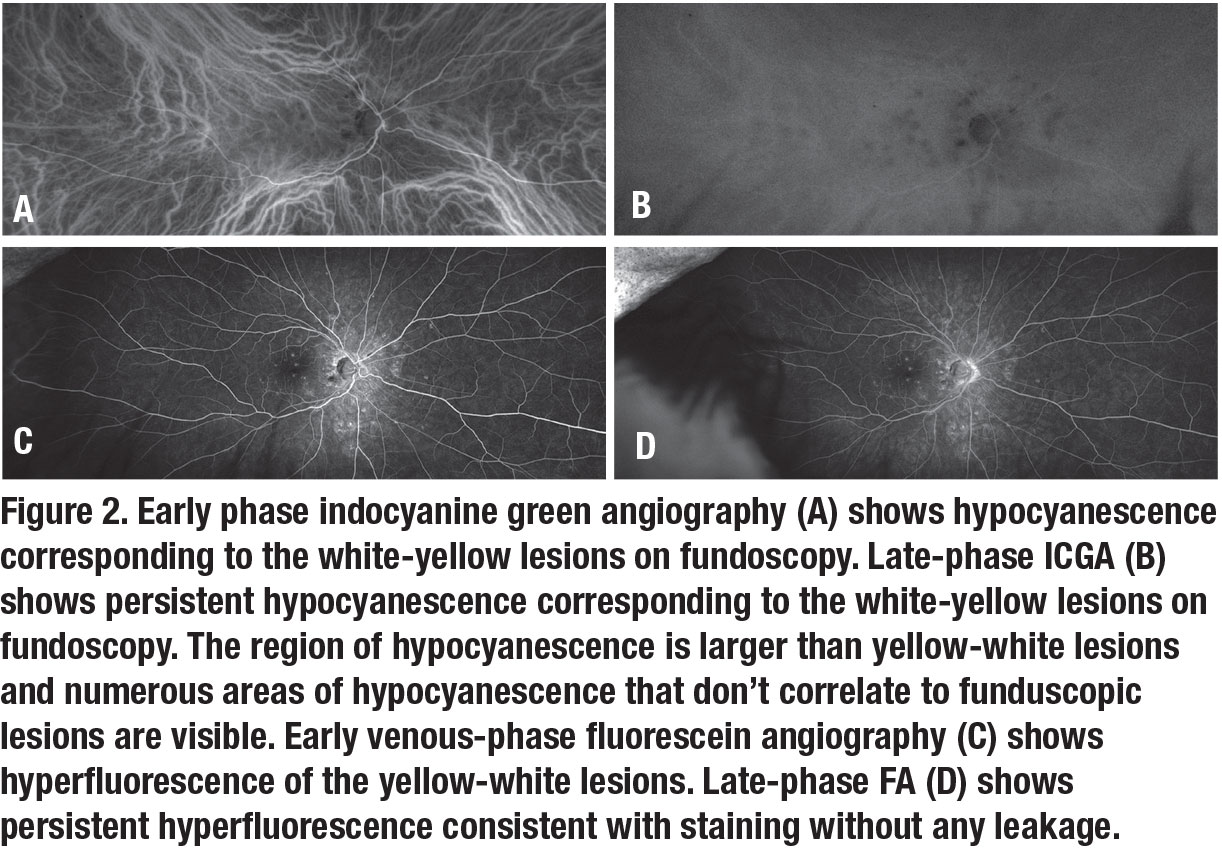
Indocyanine green angiography of the right eye demonstrated multifocal areas of early hypocyanescence (Figure 2A) that remained hypocyanescent into the late phase. While many of them corresponded to the lesions seen on funduscopy, there were additional foci of hypocyanescence. The areas of hypocyanescence were also larger than the clinical lesions. Fluorescein angiography showed early hyperfluorescence (Figure 2C) of the punctate lesions with staining in the late phase (Figure 2D).
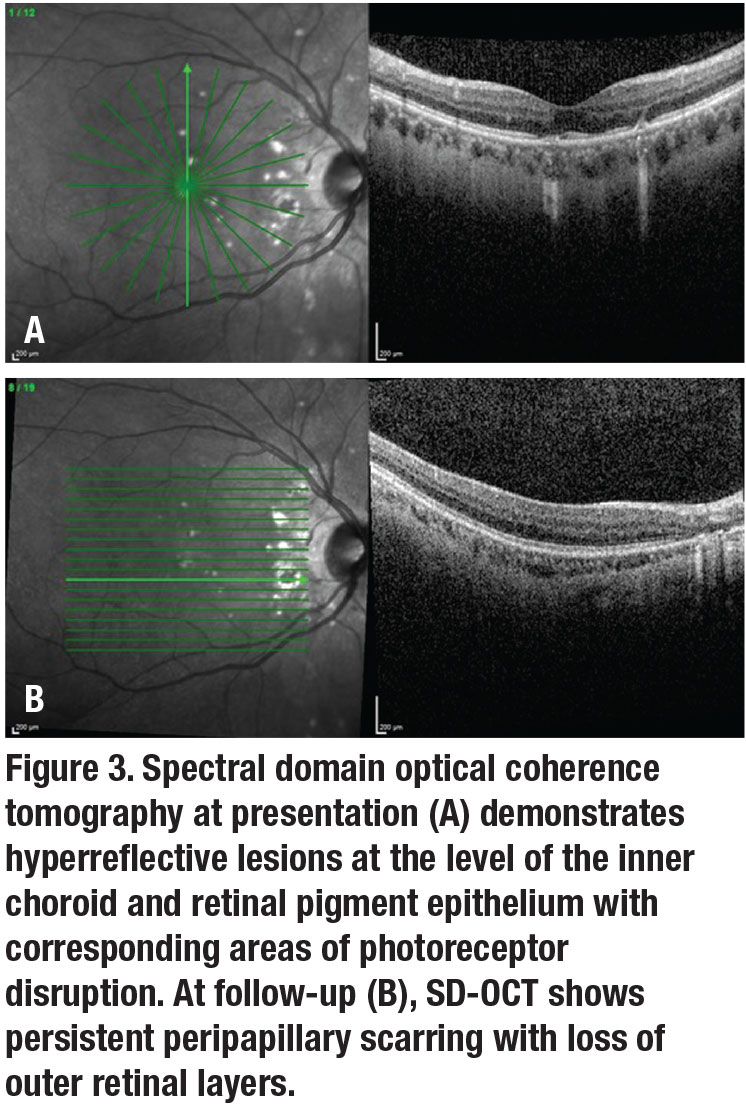
Optical coherence tomography of the right eye demonstrated deposits of moderate hyperreflectivity at the level of the inner choroid with elevation of the retinal pigment epithelium and subsequent disruption of the photoreceptor layer in the outer retina (Figure 3A, page 10). The single area of scarring revealed disruption of the RPE with atrophy of the overlying outer retinal layers (Figure 3B, page 10).
 |
Testing and diagnosis
The differential diagnosis of multifocal white dots is broad, and includes inflammatory and infectious etiologies. Possible inflammatory etiologies include:
- punctate inner choroidopathy (PIC);
- presumed ocular histoplasmosis syndrome;
- multifocal choroiditis;
- multiple evanescent white dot syndrome; and
- sarcoidosis.
We also considered possible infectious etiologies, including syphilis and tuberculosis, although these aren’t typically unilateral. Serological testing for syphilis and tuberculosis, and chest X-ray were normal. Based on the clinical presentation and classical multimodal imaging described above, we made a clinical diagnosis of PIC.
Disease presentation
Robert Watzke, MD, and colleagues first described PIC in 1984 with a cohort of 10 young myopic women.1 They reported yellow-gray lesions at the level of the inner choroid and RPE. Subsequent series reporting larger cohorts characterized the lesions as 100 to 300 µm in size with a predilection for the posterior pole in a generally random pattern.2,3 These characteristic lesions and absence of both iritis and vitritis can differentiate PIC from other white-dot syndromes.
In one series, the most common symptoms were scotoma, blurred vision, photopsia, floaters, photophobia and metamorphopsia.4 Visual acuity at presentation is variable; however, a majority of patients in retrospective studies tended to present at 20/50 or better.1–3,5 The disease can be bilateral or unilateral. The literature describes a variable rate of the former.1,3,6
The natural disease course is variable. In rare cases the lesions self-resolve. More commonly, the lesions progress to chorioretinal scarring with a “punched-out” appearance. The scars often represent areas of permanent scotomas. If the lesions involve the fovea, they can lead to a loss of central acuity. A substantial risk of choroidal neovascularization is associated with the lesions, with rates reported at 20 to 70 percent.1,3–5
 |
The diagnosis is clinical
Multimodal imaging is especially useful in aiding the diagnosis and determining the extent of the disease. OCT imaging can often confirm lesion depth at the level of the inner choroid and RPE. Breakage of the RPE is an OCT feature that often distinguishes lesions associated with permanent scarring.7
Indocyanine green angiography identifies areas of hypoperfusion at the level of the choriocapillaris and highlights lesions that are subclinical on funduscopy.7,8 Fluorescein angiography is often useful in identifying and confirming the presence of CNV.
Fundus autofluorescence is particularly helpful for assessing disease activity and treatment response. Areas of hyperautofluorescence correlate with areas of active inflammatory disease at the level of the RPE. Regression in hyperautofluorescence signifies amelioration of active inflammation either from the natural course or treatment.9
 |
Treatment
Given the risk of permanent vision loss from both the disease course and associated CNV, most retina specialists and patients elect immunosuppression once the diagnosis is established. Corticosteroids are often the first choice in treatment; both systemic and intravitreal steroids have been used. The benefit of either form is unclear based on retrospective studies, although there is suspected bias because advanced cases are more likely to receive treatment.3,4
With systemic steroids, a prednisone taper is often initiated. Steroid-sparing agents are considered if the disease relapses and the patient can’t be safely tapered off systemic steroids.10 Treatment duration is often patient- specific depending on the risks vs. benefits of long-term immunosuppression. Although controversial, some providers will initiate long-term, steroid-sparing agents on presentation.
If CNV develops, anti- VEGF agents are the first-line treatment of choice, although photodynamic therapy has been reported as an alternative. Development of CNV doesn’t necessarily signify active inflammatory disease.
In this case, we initiated oral prednisone 60 mg per day with a slow taper. Two months after presentation, the patient’s visual acuity improved to 20/20, and the photopsias and subjective scotoma had resolved.
On fundus examination, the yellow-white punctate lesions had shrunken and consolidated with sharp margins (Figure 1B, page 8). Fundus autofluorescence was notable for persistent foci of hypoautofluorescence. The rings of hyperautofluorescence, however, had resolved (Figure 1D, page 8).
Bottom line
PIC is characterized by multifocal inflammation of the inner choroid and RPE without the associated vitritis that often occurs in young, myopic females. Lesions are initially elevations at the RPE level, but areas of chronic inflammation can lead to permanent scarring with a “punched-out” appearance.
The diagnosis is clinical and multimodal imaging plays a key role in confirming and identifying areas of subclinical inflammation. Additionally, fluorescein angiography can be helpful in detecting CNV.
Treatment of the inflammatory process is often with local or systemic immunosuppression. Anti-VEGF treatment is indicated for CNV. Multimodal imaging is paramount to evaluate disease activity as well as treatment response. RS
REFERENCES
1. Watzke RC, Packer AJ, Folk JC, Benson We, Burgess D, Ober RR. Punctate inner choroidopathy. Am J Ophthalmol. 1984;98:572–584.
2. Kedhar SR, Thorne JE, Wittenberg S, Dunn JP, Jabs DA. Multifocal choroiditis with panuveitis and punctate inner choroidopathy: comparison of clinical characteristics at presentation. Retina. 2007; 27:1174–1179.
3. Essex RW, Wong J, Fraser-Bell S, et al. Punctate inner choroidopathy: clinical features and outcomes. Arch Ophthalmol. 2010; 128:982–987.
4. Gerstenblith AT, Torne JE, Sobrin L, et al. Punctate inner choroidopathy: a survey analysis of 77 persons. Ophthalmology. 2007;114:1201–1204.
5. Brown J, Folk JC, Reddy CV, Kimura AE. Visual prognosis of multifocal choroiditis, punctate inner choroidopathy, and the diffuse subretinal fibrosis syndrome. Ophthalmology. 1996;103:1100–1105.
6. Zhang X, Wen F, Zuo C, Li M, et al. Clinical features of punctate inner choroidopathy in Chinese patients. Retina. 2011;31:1680–1691.
7. Zhang X, Zuo C, Li M, Chen H, Huang Sm Wen F. Spectral-domain optical coherence tomographic findings at each stage of punctate inner choroidopathy. Ophthalmology. 2013;120:2678–2683.
8. Tiffin PAm Maini R, Roxburgh ST, Ellingford A. Indocyanine green angiography in a case of punctate inner choroidopathy. Br J Ophthalmol. 1996;80:90–91.
9. Li M, Zhang Xm Wen F. The fundus autofluorescence spectrum of punctate inner choroidopathy. J Ophthalmol. Published online July 22, 2015.
10. Turkcuoglu P, Chang PY, Rentiya ZS, et al. Mycophenolate mofetil and fundus autofluorescence in the management of recurrent punctate inner choroidopathy. Ocul Immunol Inflamm. 2011;19:286–292.



