Take-home Points
|
 |
|
Bios Dr. Lad is an associate professor at Duke University. Dr. Steinle is a partner at California Retina Consultants in Southern California. DISCLOSURES: Dr. Alekseev has no relevant relationships to disclose. Dr. Lad disclosed relationships with Apellis, Roche, Novartis, Allegro, Gallimedix, Retrotope, Gemini Therapeutics and LumiThera. Dr. Steinle disclosed relationships with Alimera Sciences, Apellis, Carl Zeiss Meditec, Genentech, Notal Vision, Novartis, Regenerative Patch Technologies, Regeneron and RegenxBio, and is an investor in Vortex Surgical. |
The complement pathway, a key component of innate immunity, maintains immune homeostasis throughout the body, including in ocular tissues. It’s composed of three pathways —classical, alternative and lectin—all eventually converging on C3 cleavage (Figure).
Endogenous inhibitory factors suppress activated complement to maintain immune equilibrium. Thus, autologous damage can arise due to overaction, whether through inappropriate activation or inadequate inhibition.
In the retina, the complement system, particularly the alternative pathway, has been implicated in the development of age-related macular degeneration and geographic atrophy. In all, six members of the complement cascade—CFB, CFH, CFI, C2, C3 and C9—are estimated to account for 40 to 60 percent of AMD heritability.1
Complement activation levels differ significantly between AMD disease stages, with higher activation corresponding with more advanced AMD and central GA.2 Many complement components and regulators deposit within drusen, where they act as inflammatory foci.3
This review examines the role of complement factors in the pathogenesis of GA.
Complement factor B
CFB is an activating factor in the alternative pathway. It’s converted by CFD into its active subunit Bb, which then becomes an integral part of the C3 convertase. Activation products of CFB (Ba and Bb) are elevated in the serum of AMD patients.3
Two protective variants of CFB have been described: R32Q and L9H.4 The R32Q polymorphism also appears to slow GA progression.5 These polymorphisms may be protective due to the impaired activity of the resultant C3 convertase.
Complement factor D
CFD is directly upstream of CFB and also activates the alternative pathway. A CFD-/- knockout mouse model of AMD showed decreased photoreceptor damage compared to wild type.6 One polymorphism (rs3826945) has been associated with AMD.7 In addition, serum levels of CFD have been shown to be elevated in AMD patients,7 a noteworthy finding since CFD is considered to be rate-limiting in the activation of the alternative pathway.
Complement factor H
CFH is a key regulatory factor responsible for complement cascade inactivation. CFH is the first and most researched complement factor to be linked to AMD.3 It prevents damage to healthy bystander cells, but not pathogens, by selective inhibition of complement activity on self-surfaces.8
Genome-wide association studies have identified numerous risk-imparting, as well as disease-protective, CFH variants. Of these, Y402H is the most notable common variant; it’s present in 50 percent of AMD patients9 and correlates with the presence of GA.10,11 This polymorphism compromises glycosaminoglycan-mediated interaction between CFH and the damaged retina, thereby attenuating inactivation of the complement cascade and allowing for chronic low-level inflammation.12
In contrast, R1210C is an example of a rare but highly penetrant variant that imparts a more severe disease course, including earlier disease onset by approximately six years.13 Interestingly, CFH accumulates in drusen at the choroid-RPE interface, where it’s thought to expose retinal pigment epithelium cells to continuous membrane attack complex (MAC) damage.9 In addition to conferring general AMD risk, CFH variants impart higher risk of conversion from large drusen to GA.14,15
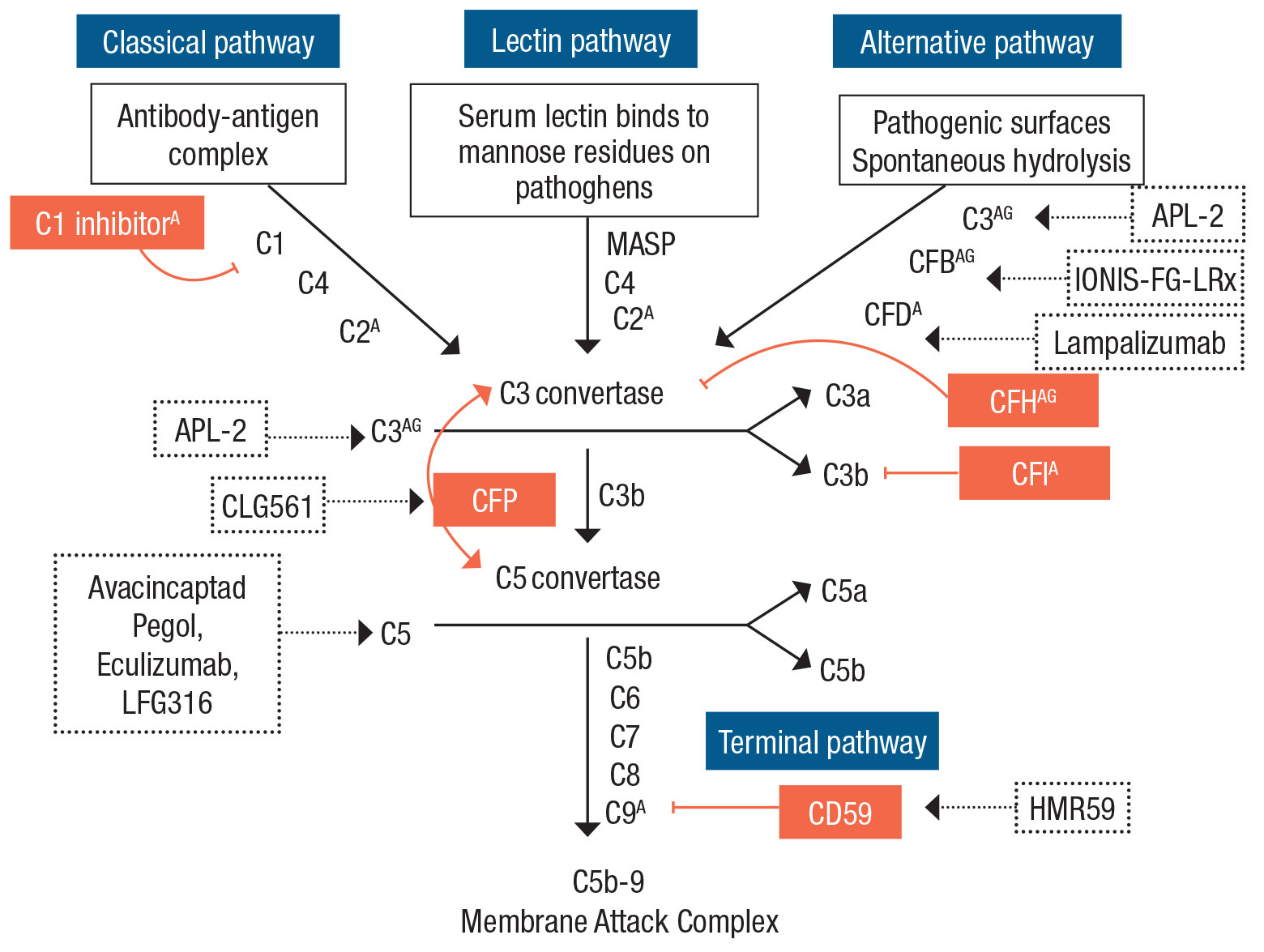 |
| Three complement pathways—classical, lectin and alternative—converge on the common terminal pathway, eventually leading to formation of the membrane attack complex. Complement factors with known genetic associations with age-related macular degeneration are marked with A (CFB, CFD, CFH, CFI, C1 inhibitor, C2, C3 and C9). Those with known genetic associations specifically with geographic atrophy are marked with G (CFB, CFH and C3). Complement regulators appear in orange boxes. Complement-targeting medications under current or former clinical trial investigation (Table, page 33) appear in dashed boxes. (Adapted from Khandhadia S, Cipriani V, Yates JR, Lotery AJ. Age-related macular degeneration and the complement system. Immunobiology. 2012;217:127-146.) |
Complement factor I
CFI is a C3b/C4b inactivator and the rate-limiting enzyme in complement termination. Interestingly, amyloid-beta, a component of drusen, binds to CFI and reduces its activity.16 While several high-risk CFI variants have been proposed, a meta-analysis has confirmed a strong association between AMD and CFI variants rs10033900T>C (protective) and rs2285714C>T (high-risk).17
Subsequently, a large systematic functional testing study revealed numerous rare CFI variants that likely contribute to AMD.18 A rare but particularly pathogenic polymorphism is G119R, which results in a hypoactive version of CFI and imparts a 20-fold increased risk of AMD.19
Complement factor P
CFP (properdin) is the only known positive regulator of the complement cascade, wherein it stabilizes the C3 and C5 convertases. While no properdin polymorphisms are known to associate with AMD, it presents an appealing therapeutic target (Table).
Complement component 1
The trigger of the classical pathway, C1 becomes activated by binding to antigen-antibody complexes. While no known AMD-causing variants of C1 have been identified, potential associations between AMD and variants in the C1-inhibitor gene, SERPING1,20 have been noted, although these findings remain contested.
Complement component 2
C2 is activated via the classical and lectin pathways to become a part of the C3 convertase. Two major protective variants of C2 are E318D and rs547154. However, since C2 and CFB are in extensive linkage disequilibrium, CFB/C2 is considered a single risk-modifying allele, wherein C2 variants likely do not have an independent molecular role in AMD.21
Complement component 3
C3 is the convergence point of the three complement pathways. It’s activated by C3 convertase to become C3a, which then becomes a core subunit of C5 convertase.
Activated C3 is often measured as a reflection of overall complement cascade activity. C3 is found deposited within drusen and elevated in plasma of AMD patients.3 C3 overexpression in mice induces retinal pathology that in many aspects recapitulates AMD, including photoreceptor and RPE atrophy.22 The most common AMD-linked C3 variants are R80G and R102G,4 whereas K155Q is a rare variant. Variant R102G is strongly associated specifically with GA.11
As a common relay point in the complement cascade, C3 is an attractive therapeutic target. A C3-neutralizing therapeutic (APL-2, also known as pegcetacoplan, Apellis Pharmaceuticals) has shown promising clinical results in a Phase II trial23 and is the subject of two fully enrolled ongoing Phase III trials24,25 (Table).
Additional agents, including a non-PEGylated compstatin (AMY-106)26 and a novel protease that cleaves and degrades C3 (CB 2782-PEG),27 are currently in preclinical development.
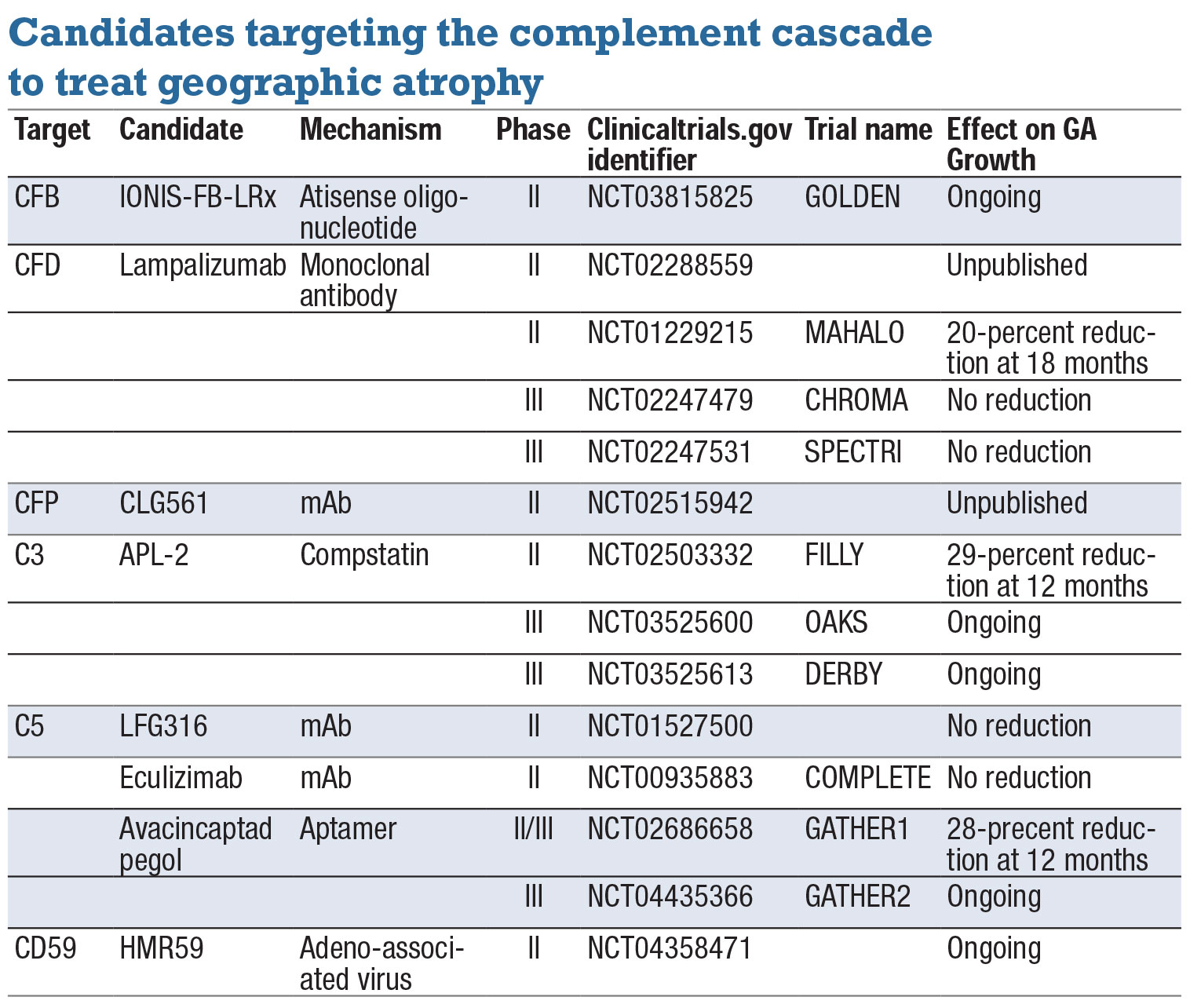 |
| To date, only three clinical trials have shown reduction in geographic atrophy growth: MAHALO, FILLY and GATHER1. Results of the Phase II MAHALO study were subsequently invalidated by the larger CHROMA and SPECTRI trials. Based on positive results of the Phase II FILLY and Phase II/III GATHER1 trials, both APL-2 and avacincaptad pegol are in Phase III clinical trials. |
Complement component 5
C5 is activated by C5 convertase to become C5a, a pro-inflammatory mediator that acts as a nidus for MAC formation. Plasma levels of C5a are elevated in AMD patients11 and stimulation of the choroid C5a receptor causes ICAM-1 overexpression, which likely leads to monocyte recruitment.28 Nevertheless, no consistent and replicable associations between polymorphisms in C5 or C5a receptors and AMD have been reported.28,29 A C5-based therapeutic (avacincaptad pegol, also known as Zimura, Iveric bio) has already shown promising Phase II/III results,30 with a second Phase III trial ongoing31 (Table).
Complement component 9
C9 participates in the final stage of MAC formation, whereby polymerization of 12-18 molecules of C9 forms a cytolytic transmembrane pore in the target cell. A high-risk C9 variant (P167S) has been reported in a mixed population of GA and/or nAMD,32 although this study didn’t stratify specifically for correlation with GA.
Another variant, R95S, has only been reported in nAMD.33 While C9 is currently not being targeted for therapeutic purposes, an endogenous inhibitor of C9 polymerization, CD59, has inspired a gene-therapy-based MAC-inhibitory agent now in a Phase II trial (Table).
Outside the pathway
 |
While most of the research into the role of complement in AMD focuses on polymorphisms that cause complement dysfunction, it’s important to recognize that the inciting pathology may lie outside the complement genes themselves. Inflammation, reactive oxygen species and complement-expressing macrophages all contribute to complement activation in AMD.
Particularly interesting is the role of iron homeostasis, critical for retinal and RPE health. Directly relevant to the pathogenesis of GA, cultured RPE cells exposed to elevated iron levels upregulate C3 gene expression, protein levels and protein secretion.34 Mice with elevated iron in the RPE develop activated C3 deposits in Bruch’s membrane.35
Moreover, iron increases retinal production and deposition of amyloid-beta,36 which is known to inhibit CFI activity.16 These findings align with the elevated iron levels in the RPE of GA-affected eyes of AMD patients37 and highlight iron-chelation as a potential approach to suppress complement activity for treatment of GA.
Bottom line
When considering the role of complement factors in GA, it’s important to recognize the broad range of outcome measures from the gene-association studies. While most of these studies examine cross-sectional AMD populations of pooled disease stages, the underlying molecular pathobiology is undoubtedly more complex. The genetic and environmental risk signature is likely quite different between the onset of early AMD, progression to GA, enlargement of GA and development of nAMD.
Relatively few studies have focused on identifying complement gene variants specifically pertinent to GA. Given that this knowledge is of utmost prognostic value and could inform the development of novel therapeutics, a focused characterization of a GA-specific genetic signature would be highly useful academically and clinically.
Phase III trials are ongoing for complement inhibition at the C3 and C5 levels.24,25,31 The results of these large trials will continue to shape how we approach GA. RS
REFERENCES
1. Fritsche LG, Fariss RN, Stambolian D, Abecasis GR, Curcio CA, Swaroop A. Age-related macular degeneration: Genetics and biology coming together. Annu Rev Genomics Hum Genet. 2014;15:151-171.
2. Heesterbeek TJ, Lechanteur YTE, Lorés-Motta L, et al. Complement activation levels are related to disease stage in AMD. Invest Ophthalmol Vis Sci. 2020;61:18.
3. Khandhadia S, Cipriani V, Yates JR, Lotery AJ. Age-related macular degeneration and the complement system. Immunobiology. 2012;217:127-146.
4. Thakkinstian A, McEvoy M, Chakravarthy U, et al. The association between complement component 2/complement factor B polymorphisms and age-related macular degeneration: A HuGE review and meta-analysis. Am J Epidemiol. 2012;176:361-372.
5. Caire J, Recalde S, Velazquez-Villoria A, et al. Growth of geographic atrophy on fundus autofluorescence and polymorphisms of CFH, CFB, C3, FHR1-3, and ARMS2 in age-related macular degeneration. JAMA Ophthamol. 2014;132:528-534.
6. Rohrer B, Guo Y, Kunchithapautham K, Gilkeson GS. Eliminating complement factor D reduces photoreceptor susceptibility to light-induced damage. Invest Ophthalmol Vis Sci. 2007;48:5282-5289.
7. Stanton CM, Yates JR, den Hollander AI, et al. Complement factor D in age-related macular degeneration. Invest Ophthalmol Vis Sci. 2011;52:8828-8834.
8. Herbert AP, Makou E, Chen ZA, et al. Complement evasion mediated by enhancement of captured factor H: implications for protection of self-surfaces from complement. J Immunol. 2015;195:4986-4998.
9. Hageman GS, Anderson DH, Johnson LV, et al. A common haplotype in the complement regulatory gene factor H (HF1/CFH) predisposes individuals to age-related macular degeneration. Proc Natl Acad Sci USA. 2005;102:7227-7232.
10. Sepp T, Khan JC, Thurlby DA, et al. Complement factor H variant Y402H is a major risk determinant for geographic atrophy and choroidal neovascularization in smokers and nonsmokers. Invest Ophthalmol Vis Sci. 2006;47:536-540.
11. Scholl HP, Fleckenstein M, Fritsche LG, et al. CFH, C3 and ARMS2 are significant risk loci for susceptibility but not for disease progression of geographic atrophy due to AMD. PloS one. 2009;4:e7418.
12. Park DH, Connor KM, Lambris JD. The challenges and promise of complement therapeutics for ocular diseases. Front Immunol. 2019;10:1007.
13. Raychaudhuri S, Iartchouk O, Chin K, et al. A rare penetrant mutation in CFH confers high risk of age-related macular degeneration. Nat Genet. 2011;43:1232-1236.
14. Seddon JM, Francis PJ, George S, Schultz DW, Rosner B, Klein ML. Association of CFH Y402H and LOC387715 A69S with progression of age-related macular degeneration. JAMA. 2007;297:1793-1800.
15. Yu Y, Reynolds R, Rosner B, Daly MJ, Seddon JM. Prospective assessment of genetic effects on progression to different stages of age-related macular degeneration using multistate Markov models. Invest Ophthalmol Vis Sci. 2012;53:1548-1556.
16. Wang J, Ohno-Matsui K, Yoshida T, et al. Altered function of factor I caused by amyloid beta: implication for pathogenesis of age-related macular degeneration from drusen. J Immunol. 2008;181:712-720.
17. Wang Q, Zhao HS, Li L. Association between complement factor I gene polymorphisms and the risk of age-related macular degeneration: A Meta-analysis of literature. Int H Ophthalmol. 2016;9:298-305.
18. Tan PL, Garrett ME, Willer JR, et al. Systematic functional testing of rare variants: Contributions of CFI to age-related macular degeneration. Invest Ophthalmol Vis Sci. 2017;58:1570-1576.
19. van de Ven JP, Nilsson SC, Tan PL, et al. A functional variant in the CFI gene confers a high risk of age-related macular degeneration. Nat Genet. 2013;45:813-817.
20. Ennis S, Jomary C, Mullins R, et al. Association between the SERPING1 gene and age-related macular degeneration: A two-stage case-control study. Lancet. 2008;372:1828-1834.
21. Gold B, Merriam JE, Zernant J, et al. Variation in factor B (BF) and complement component 2 (C2) genes is associated with age-related macular degeneration. Nat Genet. 2006;38:458-462.
22. Cashman SM, Desai A, Ramo K, Kumar-Singh R. Expression of complement component 3 (C3) from an adenovirus leads to pathology in the murine retina. Invest Ophthalmol Vis Sci. 2011;52:3436-3445.
23. Liao DS, Grossi FV, El Mehdi D, et al. Complement C3 Inhibitor pegcetacoplan for geographic atrophy secondary to age-related macular degeneration: A randomized Phase 2 trial. Ophthalmology. 2020;127:186-195.
24. Study to compare the efficacy and safety of intravitreal APL-2 therapy with sham injections in patients with geographic atrophy (GA) secondary to age-related macular degeneration. ClinicalTrials.gov Identifier: NCT03525600. https://clinicaltrials.gov/ct2/show/record/NCT03525600. Accessed December 14, 2020.
25. A study to compare the efficacy and safety of intravitreal APL-2 therapy with sham injections in patients with geographic atrophy (GA) secondary to age-related macular degeneration. ClinicalTrials.gov Identifier: NCT03525613. https://clinicaltrials.gov/ct2/show/NCT03525613. Accessed December 14, 2020.
26. Hughes S, Gumas J, Lee R, et al. Prolonged intraocular residence and retinal tissue distribution of a fourth-generation compstatin-based C3 inhibitor in non-human primates. Clin immunol. 2020;214:108391.
27. Furfine E, Rao A, Baker S, et al. Pegylated CB2782: a complement factor C3-inactivating protease and potential long-acting treatment for dry AMD. Invest Ophthalmol Vis Sci. 2019;60:374-374.
28. Skeie JM, Fingert JH, Russell SR, Stone EM, Mullins RF. Complement component C5a activates ICAM-1 expression on human choroidal endothelial cells. Invest Ophthalmol Vis Sci. 2010;51:5336-5342.
29. Baas DC, Ho L, Ennis S, et al. The complement component 5 gene and age-related macular degeneration. Ophthalmology. 2010;117:500-511.
30. Jaffe GJ, Westby K, Csaky KG, et al. C5 inhibitor avacincaptad pegol for geographic atrophy due to age-related macular degeneration: A randomized pivotal Phase 2/3 trial. Ophthalmology. Published online September 1, 2020.
31. A Phase 3 safety and efficacy study of intravitreal administration of Zimura (complement C5 inhibitor). ClinicalTrials.gov Identifier: NCT04435366. https://clinicaltrials.gov/ct2/show/NCT04435366. Accessed December 14, 2020.
32. Seddon JM, Yu Y, Miller EC, et al. Rare variants in CFI, C3 and C9 are associated with high risk of advanced age-related macular degeneration. Nat Genet. 2013;45:1366-1370.
33. Nishiguchi KM, Yasuma TR, Tomida D, et al. C9-R95X polymorphism in patients with neovascular age-related macular degeneration. Invest Ophthalmol Vis Sci. 2012;53:508-512.
34. Li Y, Song D, Song Y, et al. Iron-induced local complement component 3 (c3) up-regulation via non-canonical transforming growth factor (TGF)-β signaling in the retinal pigment epithelium. J Biol Chem. 2015;290:11918-11934.
35. Hadziahmetovic M, Dentchev T, Song Y, et al. Ceruloplasmin/hephaestin knockout mice model morphologic and molecular features of AMD. Invest Ophthalmol Vis Sci. 2008;49:2728-2736.
36. Guo LY, Alekseev O, Li Y, Song Y, Dunaief JL. Iron increases APP translation and amyloid-beta production in the retina. Exp Eye Res. 2014;129:31-37.
37. Hahn P, Milam AH, Dunaief JL. Maculas affected by age-related macular degeneration contain increased chelatable iron in the retinal pigment epithelium and Bruch’s membrane. Arch Ophthalmol. 2003;121:1099-1105.



