Take-home points
|
 |
|
Bios Dr. Thangamathesvaran is a fourth-year resident at Wilmer Eye Institute at Johns Hopkins University School of Medicine, Baltimore. Dr. Scott is an associate professor of ophthalmology at the Wilmer Eye Institute at the Johns Hopkins University School of Medicine, with a clinical research emphasis on sickle cell retinopathy. DISCLOSURES: Dr. Thangamathesvaran and Dr. Scott have no relevant disclosures. |
Sickle cell retinopathy is the most common cause of vision impairment and blindness in individuals with sickle cell disease, which affects 100,000 individuals in the United States, with a global incidence of 300,000 neonates.1
Sickle cell disease (SCD) results from a point mutation in the position six of the beta-globin gene of the hemoglobin A molecule and remains the most common inherited disorder. The abnormal hemoglobin (Hb) causes impaired tissue perfusion, resulting in multisystem end-organ damage in affected patients.
Sickle cell retinopathy (SCR) can be defined as the range of retinal vascular changes that result from repetitive vaso-occlusions and ischemia in the retinal microvasculature. SCR is stratified into nonproliferative (NPSR) and proliferative (PSR) subtypes. Sequelae of the latter are the most frequent cause of vision loss in individuals with SCD, typically due to complications from vitreous hemorrhage and/or retinal detachment.
PSR is defined as the presence of pathologic characteristic “sea fan” neovascularization that occurs from progressive retinal ischemia. Given that SCR is part of a larger systemic disease process, studies have identified hemoglobin parameters and systemic biomarkers associated with SCR development.2,3 Here, we review present management of SCR and existing evidence on how systemic therapies impact the disease.
Manifestations of SCR
Commonly observed manifestations of NPSR include salmon patch hemorrhages, black sunburst lesions (Figure 1A), macular thinning (Figure 1B), retinal vaso-occlusions and arterio-venous anastomoses. These findings can be typically monitored without intervention.
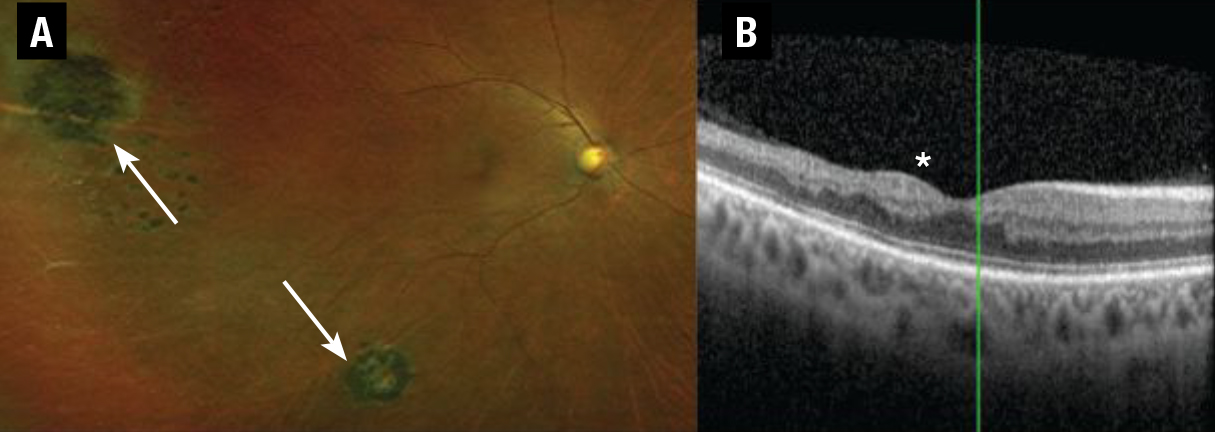 |
| Figure 1. A) Ultra-widefield fundus photograph of a patient with hemozygous sickle cell disease (HbSS) with peripheral pigmentary changes consistent with black sunburst lesions (white arrows) depicting mild nonproliferative sickle cell retinopathy (SCR). B) Spectral-domain optical coherence tomography of a patient with HbSS showing macular thinning most pronounced temporally, corresponding to the watershed zone most susceptible to ischemia in SCR (asterisk). |
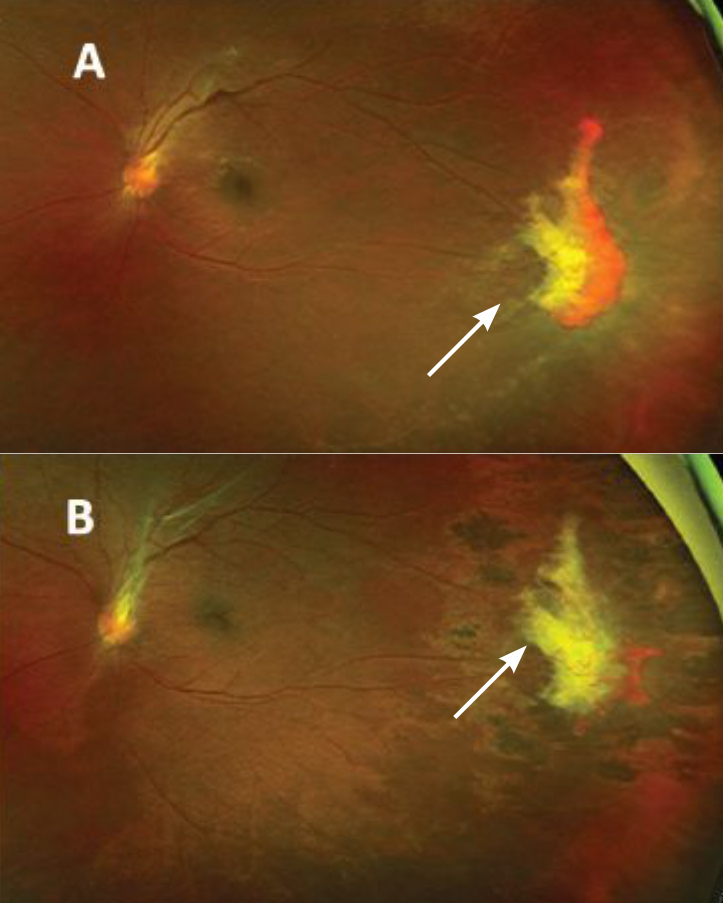
PSR can lead to visual impairment and/or blindness from vitreous hemorrhage or retinal detachment. Once PSR is detected, treatment is warranted to prevent progression to vitreous hemorrhage or retinal detachment. The gold-standard treatment for PSR remains laser photocoagulation, which barricades sea-fan neovascular complexes and targets transition zones of the peripheral retina between the perfused and nonperfused retina (Figure 2).4
Management of SCR
Although there are no currently established treatment paradigms for using
anti-VEGF therapy to treat SCR, off-label use as an adjunct to scatter laser photocoagulation has been shown to be of benefit for several indications. For example, intravitreal bevacizumab (Avastin, Genentech/Roche) has been shown to decrease vascularization of PSR complexes that remain vascularized despite scatter laser treatment.
Intravitreal bevacizumab can hasten vitreous hemorrhage resolution and it’s helpful in preventing recurrence of vitreous hemorrhage in PSR.5 Further, bevacizumab has shown utility as a preoperative medication, allowing for dissection of sea-fan neovascular complexes while minimizing intraoperative bleeding during PSR-related retinal detachment repair.6
Surgery for PSR
 |
|
Figure 2. A) Ultra-widefield fundus photography of a patient with sickle cell hemoglobin C shows peripheral retinal neovascularization representing sea-fan lesions seen in proliferative sickle cell retinopathy before treatment. B) After treatment with laser photocoagulation, the sea-fan neovascular complex shows partial regression after scatter laser treatment. |
Surgery is reserved for advanced PSR and includes non-clearing vitreous hemorrhage, retinal detachment, symptomatic epiretinal membrane, macular hole or vitreomacular traction. The surgical approach in managing patients with SCR can be complex because the peripheral retina is atrophic and thin, making it susceptible to breaks.7
Systemic agents and SCR
Systemic therapies for the management of SCR include red blood cell exchange transfusions and hydroxyurea (Hydrea, Bristol Myers Squibb). These agents reduce total sickle hemoglobin (HbS) red cells to minimize sickling and promote adequate blood flow and tissue perfusion.
Hydroxyurea, the first systemic medication approved for use in SCD, increases the production of fetal Hb (HbF). In patients with homozygous HbS disease (HbSS), studies have shown hydroxyurea slows end-organ damage in SCD and decreases the incidence of comorbidities, including vaso-occlusive pain crisis, dactylitis and acute chest syndrome.8
A study by Umar Mian, MD, and colleagues noted that the threshold for a 50- percent reduction in SCR is HbF levels of 15 percent.9 However, this and another study10 failed to stratify SCR into PSR and NPSR, limiting the conclusions that can be drawn.
Furthermore, hydroxyurea can impact SCR changes on a structural level. A prospective study led by Jennifer Lim, MD, noted decreased rates of macular thinning in patients who received hydroxyurea compared to those with SCD who didn’t receive hydroxyurea.11
Hydroxyurea may work to improve perfusion, reducing retinal ischemia and retinal thinning.11 Other work corroborated this hypothesis, documenting decreased intermittent perfusion index (IPI) on optical coherence tomography angiography two months following initiation of hydroxyurea in a patient with HbSS disease (Figure 3, below).12
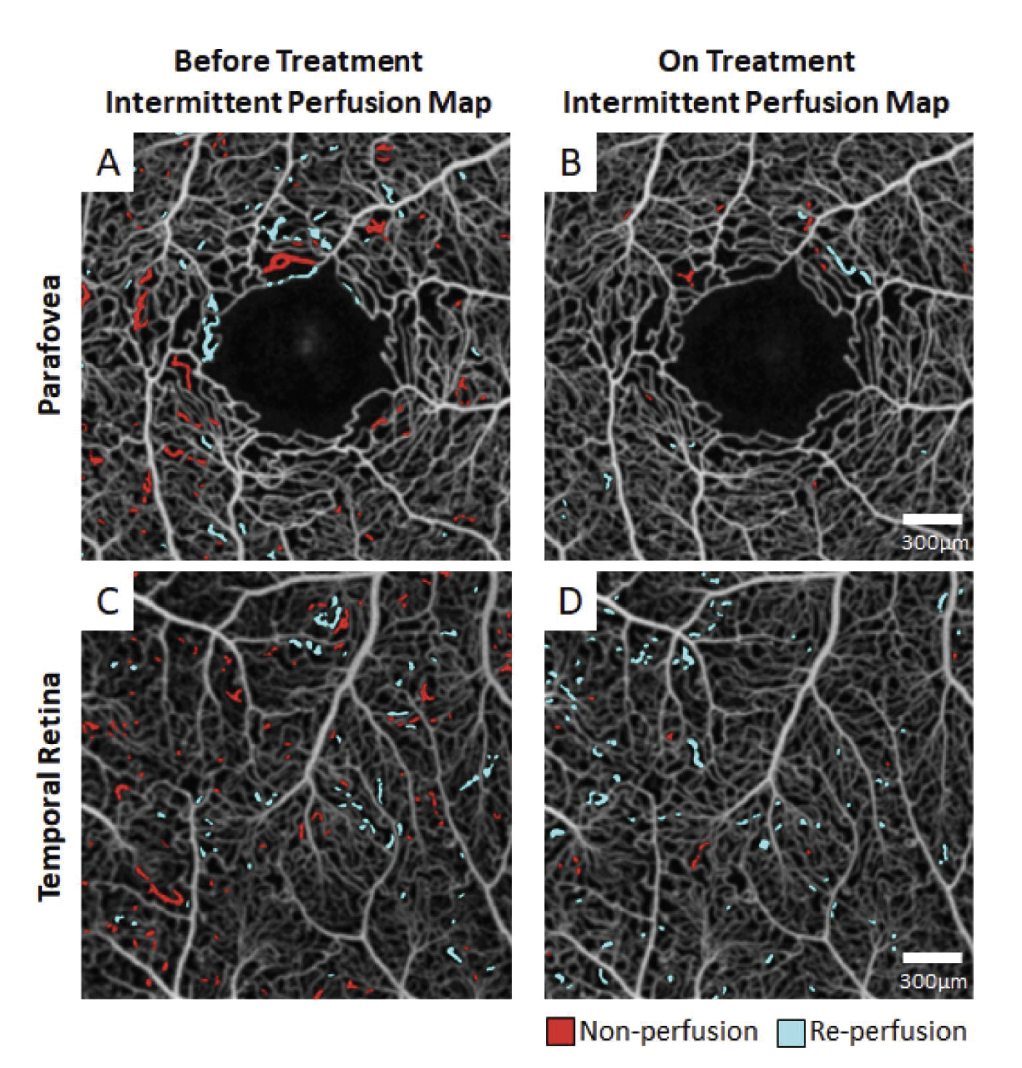 |
| Figure 3. A treatment-naive sickle cell patient with homozygous hemoglobin sickle cell disease (HbSS) genotype was imaged at an initial visit (left column) and again after two months of hydroxyurea treatment (right column). Before treatment, the patient exhibited between-session intermittent perfusion index (IPI) of 3 and 2.7 percent, respectively, at the (A) parafovea and (C) temporal retina. After two months of hydroxyurea, the between-session IPI was reduced to 0.5 and 1.6 percent, respectively, at the (B) parafovea and (D) temporal retina. (New York Eye and Ear Infirmary of Mount Sinai) |
Red blood cell exchange transfusions are routine for systemic SCD, and have decreased the frequency of vaso-occlusive pain crises, stroke and silent cerebral infarctions.13 Anecdotal case reports have demonstrated that exchange transfusions may halt PSR progression. However larger, systematic clinical studies are lacking.10,14
Systemic therapy pros and cons
Challenges with systemic use of hydroxyurea exist. Animal studies have suggested that hydroxyurea is teratogenic and can predispose young patients to developmental anomalies, including neural tube defects, digit hypoplasia and craniofacial defects. However, these findings haven’t been reproduced in human studies.15
Some patients experience neutropenia, hair loss, fingernail darkening and nausea with hydroxyurea use.16 Furthermore, hydroxyurea adherence rates as low as 30 percent have been reported. The most-cited reasons include difficulty remembering to take the medication, doubting its effectiveness and concerns about side effects.17 Nonetheless, data showing benefits of hydroxyurea significantly outweigh the reported potential side effects. One analysis reported that hydroxyurea was associated with a 40-percent reduction in mortality in patients with HbSS disease.18
Red blood cell exchange transfusions, although their role in SCR management is unclear, are used regularly in the management of systemic complications of HbSS. Indications for red cell transfusions include acute stroke, acute chest syndrome and multiorgan failure.18 These acute risks are similar to those seen with other transfusions, including the possibility of blood transmissible disease, blood clots and alterations in blood electrolytes. Risks associated with chronic transfusions include iron overload, which can be mitigated with chelation.18
Curative therapies for SCD
Hematopoietic stem cell bone marrow transplantation (BMT) and gene therapy are two novel modalities that present promising, potentially curative treatments to SCD.
The process for marrow transplant first includes chemotherapy, which is used to remove patient’s native stem cells, with busulfan being the most commonly used agent. Then the patient’s bone marrow is replaced with matched donor stem cells and the patient simultaneously receives graft versus host disease (GVHD) prophylaxis with systemic immunosuppressants to prevent rejection.
However, challenges with BMT include finding a donor match, GVHD and toxicity from immunosuppressive medications, including secondary malignancy and infertility.19
The potential of gene therapy
The second potentially curative modality is gene therapy. Gene modification mechanisms include:
- gene addition, which involves using a viral vector to deliver a nonsickling globin gene to stem cells;
- gene editing, which involves gene disruption by binding to a certain element of a gene and inducing double stranded breaks;
- gene silencing by preventing expression of certain proteins; and
- gene correction, which uses RNA as a vector to identify mutations and a template DNA to correct the mutation.
Gene therapy also carries significant potential risks, including infertility in the myeloablation process, and the potential for myelodysplastic syndromes and secondary malignancy.20 Although both BMT and gene therapy are promising, their large-scale application is limited. It will be interesting to evaluate how these modalities impact microvasculature circulation patterns and SCR progression.
Differences in SCD variants
Both HbSS and sickle cell Hb C (HbSC) disease can present with similar systemic and ocular manifestations. Although HbSS disease is associated with more severe systemic complications, vision-threatening proliferative sickle cell retinopathy is more common in HbSC disease.21
The difference in retinopathy between HbSS and HbSC genotypes isn’t completely understood. One hypothesis for increased PSR in HbSC is the partial occlusion of retinal microvasculature resulting in a chronic, steady hypoxic state promoting vaso-proliferation in contrast to the complete vaso-occlusion seen during sickling events in HbSS disease.22
Differences in pathophysiologic disease processes between HbSC and HbSS suggest that isolated studies evaluating systemic agents to prevent retinopathy in HbSC patients would be beneficial because findings from one disease process can’t be directly applied to the other.23
More PSR, macular thinning
In addition to experiencing a higher incidence of vision-threatening PSR, patients with HbSC disease also have higher rates of macular thinning than their HbSS counterparts and the lowest rates of hydroxyurea therapy.11
This difference can be attributed to current practice patterns, because hydroxyurea isn’t part of the standard systemic treatment paradigm for HbSC. Larger-scale clinical studies would help to evaluate hydroxyurea as an agent to mitigate the effects of PSR in HbSC patients.11,24
Although studies have noted that baseline Hb greater than 12.5 g/dl and HbF are risk factors for PSR, and exchange transfusions may minimize retinopathy development in HbSS, few studies in HbSC disease describe systemic interventions to prevent PSR. Similar to hydroxyurea, case reports have identified the use of transfusions to improve PSR in patients with HbSC-associated SCR, but large-scale analyses are limited.13
Future therapies
New systemic agents for SCD include:
Crizanlizumab (Adakveo, Novartis), a monoclonal antibody that inhibits the interaction of P-selectin glycoprotein ligand and thrombin reducing vaso-occlusion and thrombosis. Preliminary randomized controlled trial results noted lower crisis events and decreased median time to the first crisis with its use.25
Voxelotor (Oxbryta, Global Blood Therapeutics), a HbS polymerization inhibitor that reversibly binds to Hb to stabilize the oxygenated Hb state. Phase III randomized trials have shown increased Hb levels and reduced hemolysis markers in SCD patients.26
Amino acid L-glutamine (Endari, Emmaus Medical), a pharmaceutical grade 1 glutamine that increases the proportion of nicotinamide adenine dinucleotide, an antioxidant molecule that, in a multicenter, Phase III trial has been shown to reduce the median number of pain crises.27
Although these agents have yet to be evaluated in SCR, their systemic benefits suggest possible benefits for retinal perfusion, with potential alterations in future SCR treatment paradigms.28
Bottom line
SCR is the most common cause of vision impairment in people with SCD, and while laser photocoagulation is the gold standard for PSR, off-label bevacizumab has become a useful adjunct. Systemic therapies for SCD have shown some clinical signals to improve SCR, but they also bring limitations, as do BMT and gene therapy. The impact that newer therapies have on SCR has yet to be elucidated. RS
References
1. Wastnedge E, Waters D, Patel S, et al. The global burden of sickle cell disease in children under five years of age: A systematic review and meta-analysis. J Glob Health. 2018;8:021103. doi:10.7189/jogh.08.021103
2. Dell'Arti L, Barteselli G, Riva L, et al. Sickle cell maculopathy: Identification of systemic risk factors, and microstructural analysis of individual retinal layers of the macula. PLoS One. 2018;13:e0193582.
3. Duan XJ, Lanzkron S, Linz MO, Ewing C, Wang J, Scott AW. Clinical and ophthalmic factors associated with the severity of sickle cell retinopathy. Am J Ophthalmol. 2019;197:105-113.
4. Rodrigues M, Kashiwabuchi F, Deshpande M, et al. Expression pattern of HIF-1α and VEGF supports circumferential application of scatter laser for proliferative sickle retinopathy. Invest Ophthalmol Vis Sci. 2016;57:6739-6746.
5. Cai CX, Linz MO, Scott AW. Intravitreal bevacizumab for proliferative sickle retinopathy: a case series. J VitreoRetinal Dis. 2018;2:32-38.
6. Moshiri A, Ha NK, Ko FS, Scott AW. Bevacizumab presurgical treatment for proliferative sickle-cell retinopathy-related retinal detachment. Retin Cases Brief Rep. 2013;7:204-205.
7. Chen RW, Flynn HW, Jr., Lee WH, et al. Vitreoretinal management and surgical outcomes in proliferative sickle retinopathy: a case series. Am J Ophthalmol. 2014;157:870-875.
8. Wang WC, Ware RE, Miller ST, et al. Hydroxycarbamide in very young children with sickle-cell anaemia: A multicentre, randomised, controlled trial (BABY HUG). Lancet. 2011;377:1663-1672.
9. Mian UK, Tang J, Allende APM, et al. Elevated fetal haemoglobin levels are associated with decreased incidence of retinopathy in adults with sickle cell disease. Br J Haematol. 2018;183:807-811.
10. Agrawal RK, Patel RK, Shah V, Nainiwal L, Trivedi B. Hydroxyurea in sickle cell disease: drug review. Indian J Hematol Blood Transfus. 2014;30:91-96.
11. Lim JI, Niec M, Sun J, Cao D. Longitudinal assessment of retinal thinning in adults with and without sickle cell retinopathy using spectral-domain optical coherence tomography. JAMA Ophthalmol. 2021;139:330-337.
12. Zhou DB, Castanos MV, Pinhas A, et al. Quantification of intermittent retinal capillary perfusion in sickle cell disease. Biomed Opt Express. 2021;12:2825-2840.
13. Howard J. Sickle cell disease: when and how to transfuse. Hematol Am Soc Hematol Educ Program. 2016;2016:625-631.
14. McKinney CM, Siringo F, Olson JL, Capocelli KE, Ambruso DR, Nuss R. Red cell exchange transfusion halts progressive proliferative sickle cell retinopathy in a teenaged patient with hemoglobin SC disease. Pediatr Blood Cancer. 2015;62:721-723.
15. Schlisser AE, Hales BF. Deprenyl enhances the teratogenicity of hydroxyurea in organogenesis stage mouse embryos. Toxicol Sci. 2013;134:391-399.
16. Weber GF. DNA Damaging Drugs. In: Weber GF, ed. Molecular Therapies of Cancer. Springer International Publishing; 2015:9-112.
17. Hodges JR, Phillips SM, Norell S, et al. Intentional and unintentional nonadherence to hydroxyurea among people with sickle cell disease: a qualitative study. Blood Advances. 2020;4:4463-4473.
18. Steinberg MH, Barton F, Castro O, et al. Effect of hydroxyurea on mortality and morbidity in adult sickle cell anemia: risks and benefits up to 9 years of treatment. JAMA. 2003;289:1645-1651.
19. Ashorobi D, Bhatt R. Bone Marrow Transplantation In Sickle Cell Disease. StatPearls. StatPearls Publishing LLC.; 2022.
20. Kanter J, Falcon C. Gene therapy for sickle cell disease: Where we are now? Hematology. 2021;2021:174-180.
21. Fox PD, Dunn DT, Morris JS, Serjeant GR. Risk factors for proliferative sickle retinopathy. Br J Ophthalmol. 1990;74:172-176. doi:10.1136/bjo.74.3.172
22. Ballas SK, Lewis CN, Noone AM, Krasnow SH, Kamarulzaman E, Burka ER. Clinical, hematological, and biochemical features of Hb SC disease. Am J Hematol. 1982;13:37-51.
23. Hingorani M, Bentley CR, Jackson H, et al. Retinopathy in haemoglobin C trait. Eye (Lond). 1996;10:338-342.
24. Summarell CCG, Sheehan VA. Original Research: Use of hydroxyurea and phlebotomy in pediatric patients with hemoglobin SC disease. Exp Biol Med (Maywood). 2016;241:737-744.
25. Ataga KI, Kutlar A, Kanter J, et al. Crizanlizumab for the prevention of pain crises in sickle cell disease. N Engl J Med. 2017;376:429-439.
26. Vichinsky E, Hoppe CC, Ataga KI, et al. A Phase 3 randomized trial of voxelotor in sickle cell disease. N Engl J Med. 2019;381:509-519.
27. Niihara Y, Miller ST, Kanter J, et al. A Phase 3 trial of l-glutamine in sickle cell disease. N Engl J Med. 2018;379:226-235.
28. Wu Y, Zeng J, Roscoe BP, et al. Highly efficient therapeutic gene editing of human hematopoietic stem cells. Nat Med. 2019;25:776-783.



