 |
 |
 |
 |
Inherited retinal diseases affect approximately 200,000 people in the United States and 4.5 million people worldwide.1 Most IRDs are caused by mutations in single genes that may be amenable to gene therapy. However, while IRDs have been historically classified based on clinical features and electroretinography, genetic testing has revealed that many different genetic mutations can cause each IRD. Because most current gene therapies target a specific gene or gene mutation, the heterogeneity of IRDs requires a wide array of different treatment strategies to be developed.
Compared to pharmacologic agents, gene therapies have the potential advantages of long-term therapeutic effects and capacity for cell-targeted therapy; for example, by using cell-specific promotors. The term “gene therapy” typically refers to gene-replacement therapy, where a normal, functional copy of a gene is introduced to replace the mutated gene. However, gene-replacement therapies are useful mainly for recessive IRDs, where neither of the two mutated alleles can produce functional gene products.
By contrast, dominant IRDs are not amenable to gene replacement because the mutant gene product interacts or interferes with normal protein function, even if an exogenous source produces it. Instead, dominant IRDs must be treated by inactivating the mutant protein or ablating the mutant gene at the DNA level; for example, by using CRISPR-based
genome editing.
Gene-delivery strategies
Both gene-replacement and genome-editing therapies require the transfer of genetic materials to host cells, which can be accomplished using viral or non-viral methods. Many viruses have naturally evolved to infect human cells, and can be engineered to carry a therapeutic transgene. The desired characteristics of gene-delivery vectors include high expression, long durability, large capacity, low immunogenicity and low risk of mutagenicity (Table 1). The important gene-delivery vectors are:
- Lentiviruses, which can deliver approximately 8-kilobase (kb)-long DNA sequences for long-term expression, but this process has a mild risk of mutagenesis because it integrates into the host genome.
- Adenoviruses, which do not integrate, but are limited by their short durability and high immunogenicity that can cause uveitis.
- Adeno-associated viruses (AAVs), the most common platform because they are non-pathogenic and non-
integrating. AAVs with different capsid serotypes can target different cell types, with intravitreal AAV2 and AAV8 infecting retinal ganglion cells, and subretinal AAV2, AAV5, AAV7, AAV8 and AAV9 transducing photoreceptors and RPE.2–5 AAVs have a limited carrying capacity of approximately 4.7 kb, so larger genes such as the ABCA4 gene for Stargardt disease cannot be delivered using a single AAV vector. - Non-viral approaches, such as synthetic polymers and nanoparticles, are generally safe, but are not as effective for gene transfer as viruses.
 |
Delivering vectors
Vectors are generally delivered by subretinal or intravitreal injections. Intravitreal injections are easier to perform, but most AAV cannot penetrate the internal limiting membrane to reach the outer retina. Subretinal injections can overcome the ILM barrier, but they require invasive vitrectomy surgery and the therapeutic effect is limited to the small area of the subretinal bleb.
Newer generations of AAV developed by “directed evolution” have shown promise in crossing the ILM from the vitreous, and are in clinical trials. Our laboratory recently demonstrated the possibility of suprachoroidal AAV delivery using
micro-needles, which provide widespread but mostly peripheral transgene expression that may be useful for “biofactory” therapies such as production of anti-
angiogenic factors. Compared with intravitreal and suprachoroidal delivery, however, the subretinal space is immune-privileged and is less prone to triggering intraocular inflammation.5
 |
Gene-replacement therapies
 |
• Type 2 Leber congenital amaurosis. The best-known ocular gene therapy is for the treatment of type 2 LCA. LCA is an autosomal recessive IRD that occurs in one in 80,000 births, and is associated with mutations in the GUCY2D, CEP290 and RPE65 genes.6 RPE65 is involved in the production of 11-cis retinal during phototransduction, and accounts for 5 to 10 percent of LCA cases.7 In animal models including RPE65-deficient mice and dogs, subretinal AAV2-mediated RPE65 delivery improved visual function8-10 and, despite relatively modest efficacy in early human trials,11,12 demonstrated safety and benefit in Phase III studies which led to its approval by the Food and Drug Administration in 2017 (Luxturna, Spark Therapeutics).13
• Choroideremia. This X-linked recessive IRD occurs in one in 50,000 males.14 Symptoms include night blindness and gradual vision loss beginning in childhood. The CHM gene encodes the Rab escort protein 1 (REP1) essential for intracellular vesicular transport.15 Subretinal AAV2-REP1 in CHM knockout mice demonstrated improvements in electroretinogram,16 and Phase I/II trials have shown improved visual acuity in some patients with no significant adverse effects.17,18 Phase II trials began in 2015 and are ongoing in patients with early choroideremia.
• Stargardt disease. This autosomal recessive macular atrophy has a prevalence of one in 10,000 people.19 The disease-causing ABCA4 gene encodes a transmembrane protein in photoreceptor outer segments, and its dysfunction results in accumulation of toxic A2E byproducts that accumulate in the retinal pigment epithelium leading to photoreceptor death.20
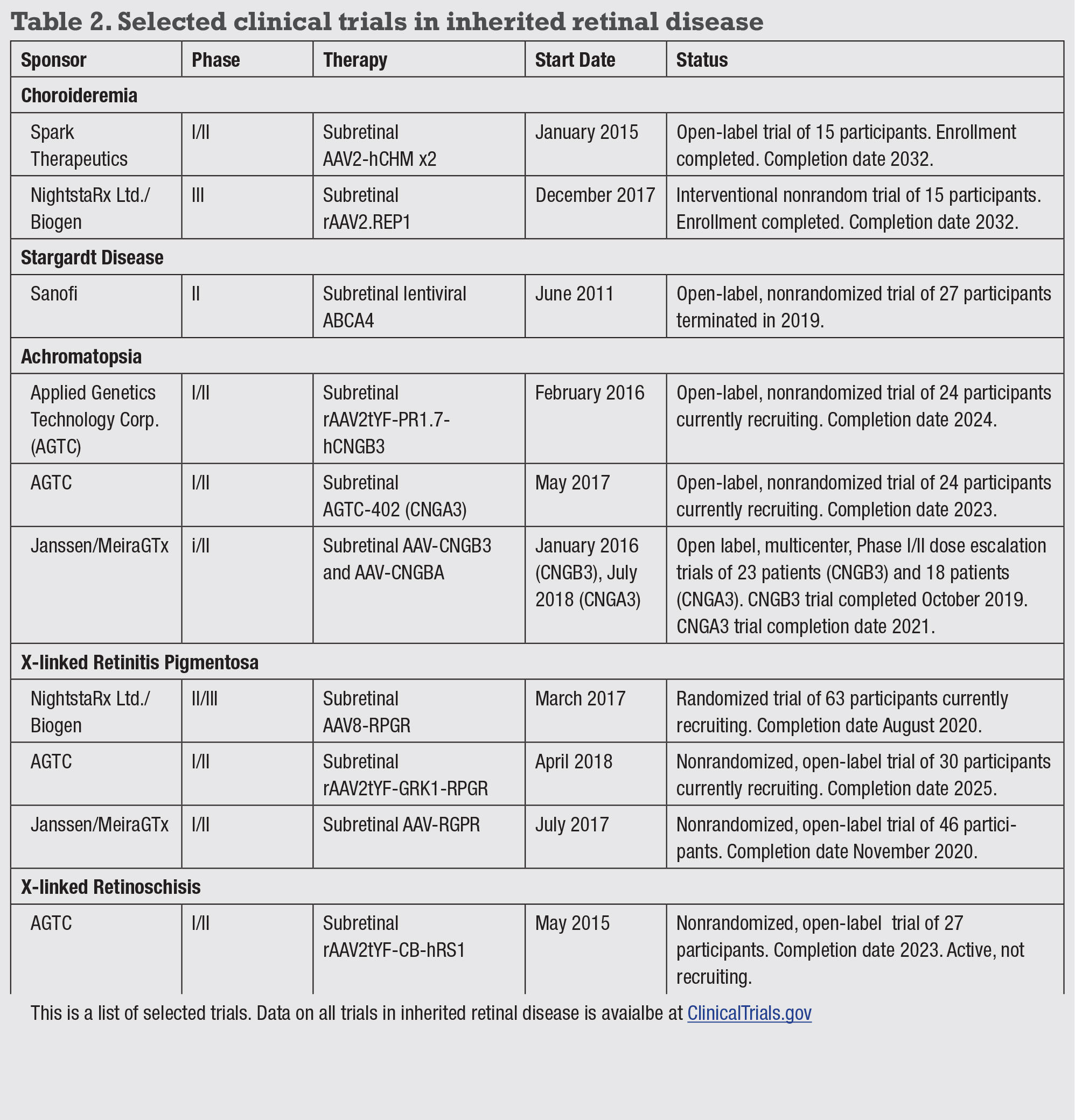
Nonsense mutations cause early onset disease in childhood with more severe atrophy, whereas missense variants are usually adult-onset, often sparing the fovea.21 The current Phase II trial employs a lentiviral vector due to the large size of the ABCA4 gene,22 although preclinical studies using a hybrid dual AAV system also show promise in ABCA-/- mice.23(p4) Gene therapies for many other IRDs are under development (Table 2).
 |
 |
CRISPR-based genome editing
Unlike conventional gene replacement therapy, genome editing using Clustered Regularly Interspaced Palindromic Repeats (CRISPR) technology enables treatment of dominant IRDs by targeting genetic mutations at the DNA level. Unlike previous genome surgery tools like TALEN or zinc finger nuclei, which are time-consuming and expensive to develop, CRISPR-based treatments can be quickly designed and easily generated.
CRISPR systems were first discovered in prokaryotes as part of their adaptive immune system against viral invasion. Bacteria infected by bacteriophages incorporate viral DNA fragments into their genome, which can be transcribed to guide RNA (gRNA) to program CRISPR-associated endonucleases, such as Cas9, to cut these foreign DNA sequences upon future encounters.
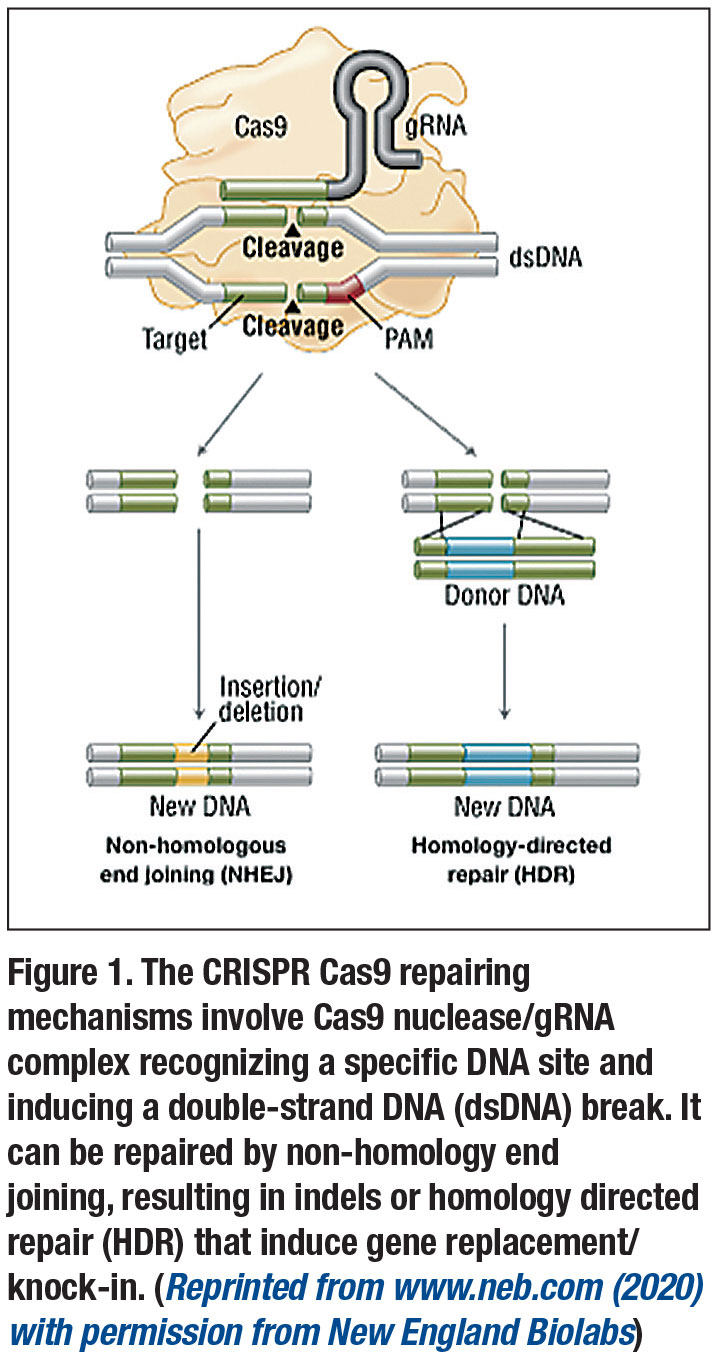
By designing gRNAs to program Cas9 enzymes to precisely cut a target gene locus,24,25 CRISPR can be used to:
- ablate a normal gene or destroy a dominant mutant allele by allowing natural, error-prone DNA repair processes that result in insertion or deletion of several nucleotides to cause frameshift mutations (indels); or
- repair a mutated gene by providing a DNA template of the normal gene that can be incorporated through a process known as homology directed repair (HDR) (Figure 1, page 23). Current applications are mostly limited to gene ablation, as HDR is much less efficient.
 |
Early results of CRISPR genome editing
Use of CRISPR-based genome editing in IRDs has demonstrated success in animal models. One approach is by deactivating the rhodopsin mutations in rodent models of autosomal dominant retinitis pigmentosa.26–28 Another is by destroying the dominant mutation in the CEP290 gene in LCA type 10 models.29–31
Subretinal AAV delivery of this CRISPR-based strategy for LCA10 is being evaluated in Phase I/II studies. Given the heterogeneity of different disease-causing mutations, another promising approach is an “ablate-and-replace” strategy that couples CRISPR-based ablation of the endogenous rhodopsin gene in a mutation-independent manner with exogenous expression of the wild-type rhodopsin.28
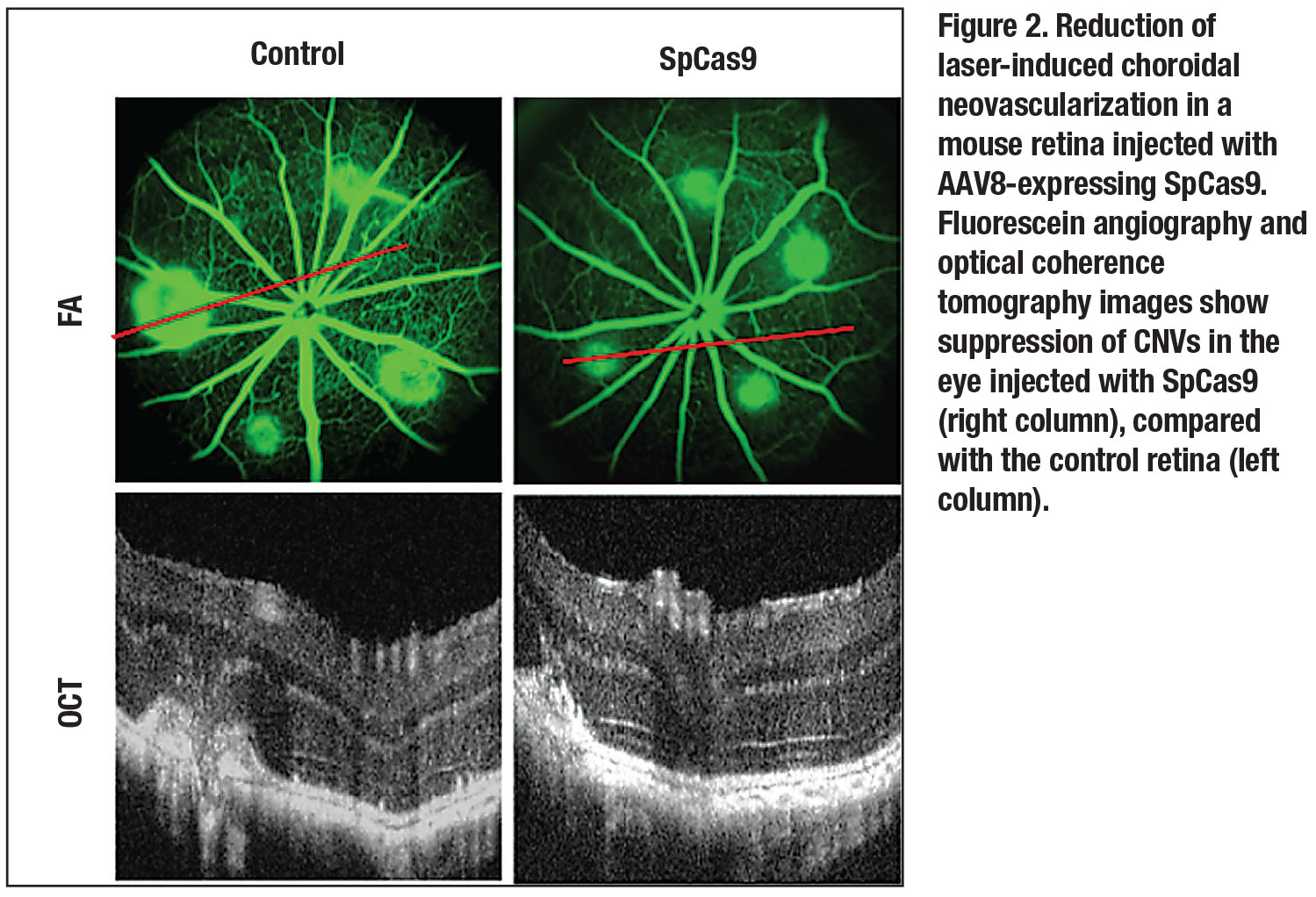
Our laboratory has also been investigating the use of CRISPR for neovascular AMD, since genomic ablation of the VEGFA gene would provide a permanent cure that contrasts with the frequent and costly anti-VEGF treatments that are currently in wide use. We first demonstrated the use of CRISPR-Cas9 to suppress VEGF from human cells in vitro,32 and more recently, in mouse models of choroidal neovascularization (CNV) in vivo (Figure 3).33
 |
 |
Bottom line
Gene replacement therapy has shown some success for recessive IRDs, with the first approved therapy for type 2 LCA and many other trials underway. Advances in gene delivery, such as new generations of AAV and suprachoroidal delivery, may further improve effectiveness. By manipulating the genome at the DNA level, CRISPR technology holds the promise of permanently curing dominant IRDs, although it has only shown success in animal studies.
However, given the bacterial origin of CRISPR systems and the permanent impact of genome editing, potential host immune responses and off-target effects require thorough investigation before human trials commence. RS
REFERENCES
1. Benati D, Patrizi C, Recchia A. Gene editing prospects for treating inherited retinal diseases. J Med Genet. Published online December 19, 2019. doi: 10.1136/jmedgenet-2019-106473.
2. Lotery AJ, Yang GS, Mullins RF, et al. Adeno-associated virus type 5: transduction efficiency and cell-type specificity in the primate retina. Hum Gene Ther. 2003;14:1663-1671.
3. Burnight ER, Giacalone JC, Cooke JA, et al. CRISPR-Cas9 genome engineering: Treating inherited retinal degeneration. Prog Retin Eye Res. 2018;65:28-49.
4. Auricchio A, Kobinger G, Anand V, et al. Exchange of surface proteins impacts on viral vector cellular specificity and transduction characteristics: The retina as a model. Hum Mol Genet. 2001;10:3075-3081.
5. Yiu G, Chung SH, Mollhoff IN, et al. Suprachoroidal and subretinal injections of AAV using transscleral microneedles for retinal gene delivery in nonhuman primates. Mol Ther Methods Clin Dev. 2020;16:179-191.
6. Tsang SH, Sharma T. Leber congenital amaurosis. Adv Exp Med Biol. 2018;1085:131-137.
7. den Hollander AI, Roepman R, Koenekoop RK, Cremers FPM. Leber congenital amaurosis: genes, proteins and disease mechanisms. Prog Retin Eye Res. 2008;27:391-419.
8. Veske A, Nilsson SE, Narfström K, Gal A. Retinal dystrophy of Swedish briard/briard-beagle dogs is due to a 4-bp deletion in RPE65. Genomics. 1999;57:57-61.
9. Cideciyan AV, Jacobson SG, Beltran WA, et al. Human retinal gene therapy for Leber congenital amaurosis shows advancing retinal degeneration despite enduring visual improvement. Proc Natl Acad Sci USA. 2013;110:E517-525.
10. Roman AJ, Boye SL, Aleman TS, et al. Electroretinographic analyses of Rpe65-mutant rd12 mice: Developing an in vivo bioassay for human gene therapy trials of Leber congenital amaurosis. Mol Vis. 2007;13:1701-1710.
11. Jacobson SG, Cideciyan AV, Roman AJ, et al. Improvement and decline in vision with gene therapy in childhood blindness. N Engl J Med. 2015;372:1920-1926.
12. Weleber RG, Pennesi ME, Wilson DJ, et al. Results at 2 years after gene therapy for RPE65-deficient Leber congenital amaurosis and severe early-childhood-onset retinal dystrophy. Ophthalmology. 2016;123:1606-1620.
13. Russell S, Bennett J, Wellman JA, et al. Efficacy and safety of voretigene neparvovec (AAV2-hRPE65v2) in patients with RPE65-mediated inherited retinal dystrophy: A randomized, controlled, open-label, Phase 3 trial. Lancet. 2017;390:849-860.
14. Xue K, MacLaren RE. Ocular gene therapy for choroideremia: clinical trials and future perspectives. Expert Rev Ophthalmol. 2018;13:129-138.
15. Larijani B, Hume AN, Tarafder AK, Seabra MC. Multiple factors contribute to inefficient prenylation of Rab27a in Rab prenylation diseases. J Biol Chem. 2003;278:46798-46804.
16. Tolmachova T, Tolmachov OE, Barnard AR, et al. Functional expression of Rab escort protein 1 following AAV2-mediated gene delivery in the retina of choroideremia mice and human cells ex vivo. J Mol Med Berl Ger. 2013;91:825-837.
17. MacLaren RE, Groppe M, Barnard AR, et al. Retinal gene therapy in patients with choroideremia: Initial findings from a Phase 1/2 clinical trial. Lancet. 2014;383:1129-1137.
18. Edwards TL, Jolly JK, Groppe M, et al. Visual acuity after retinal gene therapy for choroideremia. N Engl J Med. 2016;374:1996-1998.
19. Lambertus S, van Huet RAC, Bax NM, et al. Early-onset stargardt disease: Phenotypic and genotypic characteristics. Ophthalmology. 2015;122:335-344.
20. Tsybovsky Y, Molday RS, Palczewski K. The ATP-binding cassette transporter abca4: Structural and functional properties and role in retinal disease. Adv Exp Med Biol. 2010;703:105-125.
21. Tanna P, Strauss RW, Fujinami K, Michaelides M. Stargardt disease: linical features, molecular genetics, animal models and therapeutic options. Br J Ophthalmol. 2017;101:25-30.
22. Han Z, Conley SM, Naash MI. Gene therapy for Stargardt disease associated with ABCA4 gene. Adv Exp Med Biol. 2014;801:719-724.
23. Dyka FM, Molday LL, Chiodo VA, Molday RS, Hauswirth WW. Dual ABCA4-AAV vector treatment reduces pathogenic retinal A2E accumulation in a mouse model of autosomal recessive Stargardt disease. Hum Gene Ther. 2019;30:1361-1370.
24. Ran FA, Hsu PD, Wright J, Agarwala V, Scott DA, Zhang F. Genome engineering using the CRISPR-Cas9 system. Nat Protoc. 2013;8:2281-2308.
25. Jinek M, Chylinski K, Fonfara I, Hauer M, Doudna JA, Charpentier E. A programmable dual-RNA-guided DNA endonuclease in adaptive bacterial immunity. Science. 2012;337:816-821.
26. Latella MC, Di Salvo MT, Cocchiarella F, et al. In vivo editing of the human mutant rhodopsin gene by electroporation of plasmid-based CRISPR/Cas9 in the mouse retina. Mol Ther Nucleic Acids. 2016;5:e389.
27. Bakondi B, Lv W, Lu B, et al. In Vivo CRISPR/Cas9 Gene editing corrects retinal dystrophy in the S334ter-3 rat model of autosomal dominant retinitis pigmentosa. Mol Ther J Am Soc Gene Ther. 2016;24:556-563.
28. Tsai Y-T, Wu W-H, Lee T-T, et al. Clustered regularly interspaced short palindromic repeats-based genome surgery for the treatment of autosomal dominant retinitis pigmentosa. Ophthalmology. 2018;125:1421-1430.
29. den Hollander AI, Koenekoop RK, Yzer S, et al. Mutations in the CEP290 (NPHP6) gene are a frequent cause of Leber congenital amaurosis. Am J Hum Genet. 2006;79:556-561.
30. Ruan G-X, Barry E, Yu D, Lukason M, Cheng SH, Scaria A. CRISPR/Cas9-mediated genome editing as a therapeutic approach for Leber congenital amaurosis 10. Mol Ther J Am Soc Gene Ther. 2017;25:331-341.
31. Maeder ML, Stefanidakis M, Wilson CJ, et al. Development of a gene-editing approach to restore vision loss in Leber congenital amaurosis type 10. Nat Med. 2019;25:229-233.
32. Yiu G, Tieu E, Nguyen AT, Wong B, Smit-McBride Z. Genomic disruption of VEGF-a expression in human retinal pigment epithelial cells using CRISPR-Cas9 endonuclease. Investig Opthalmology Vis Sci. 2016;57:5490.
33. Chung SH, Mollhoff IN, Nguyen U, et al. Factors impacting efficacy of AAV-mediated CRISPR-based genome editing for treatment of choroidal neovascularization. Mol Ther Methods Clin Dev. Published online January 23, 2020. doi:https://doi.org/10.1016/j.omtm.2020.01.006



