 |
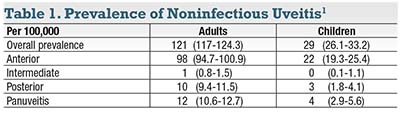
Noninfectious uveitis affects approximately 300,000 American adults and 22,000 American children (Table 1).1 Many of these patients are young, and under-treatment early in the disease course can lead to a poor visual outcome that burdens them for decades. In this article, I present a simple yet powerful therapeutic strategy that the retina specialist who does not have a fellowship in uveitis can utilize.
 |
Diagnostic Considerations
Syphilis testing (commonly Treponema pallidum IgG antibodies)2,3 and QuantiFERON testing are recommended for all uveitis patients, as well as a chest X-ray. Additional testing is tailored to the individual clinical presentation. Some specialists say that routine testing for tuberculosis is not indicated if the exam findings are not suggestive (such as serpiginous lesions, choroidal granuloma or occlusive vasculitis). However, QuantiFERON testing is still worthwhile as a precondition for any contemplated systemic immunomodulatory therapy.
If, after appropriate diagnostic testing, a concern lingers for a cryptic infection (such as endogenous fungal endophthalmitis or atypical toxoplasmosis), consider a three-to-six-week trial of oral corticosteroids before any local (periocular or intravitreal) injected corticosteroid. Follow the patient especially closely in the first two weeks of therapy. A worsening of symptoms after the patient starts oral steroids is a signal to revisit the possibility of an infectious uveitis.
If the patient passes
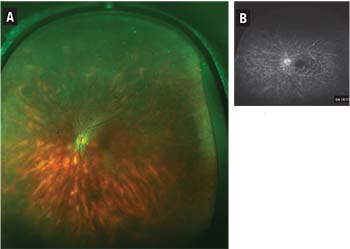 |
| Figure 1. Color fundus photograph of bird-shot retinochoroiditis in a 62-year-old woman (A) and a late-phase fluorescein angiogram of the same eye (B). Courtesy Vicky Wolzen, CRA, COA, Dean McGee Eye Institute |
Why Systemic Therapy?
Retina specialists have many good options for local therapy, including periocular triamcinolone, intravitreal triamcinolone, the dexamethasone intravitreal implant (Ozurdex, Allergan), and the fluocinolone acetonide 0.59-mg intravitreal implant (Retisert, Bausch +Lomb).
In addition,
 |
Many patients have bilateral disease, and keeping these eyes out of the operating room is a laudable goal. Repeated local steroid therapy over the course of years often provokes cataract and glaucoma. Subsequently, surgery for cataract and glaucoma may actually worsen inflammation and lead to anatomic sequelae (synechiae, fibrin deposits on an intraocular lens, anterior vitreous fibrosis and opacity, or macular edema) that further degrade vision—a virtual death spiral of uveitic complications and frustration.
Lessons from MUST Trial
Data from the Multicenter Uveitis Steroid Treatment (MUST) Trial deserves attention. Two arms of the study compared the fluocinolone acetonide implant to systemic immunomodulatory therapy. At 4.5 years, visual acuity outcomes were similar between the two arms.6,7 Systemic complications were minimal and equivalent between the two arms, but the implant arm showed significant local complications, including cataract, ocular hypertension and need for glaucoma surgery.
Especially for bilateral disease, systemic therapy may be a better first choice given its efficacy and systemic and ocular safety. Recently reported seven-year extended follow-up showed an average 7.2-letter visual acuity advantage of systemic therapy vs. the fluocinolone implant.8
 |
From a lifestyle perspective, patients often prefer a regimen of a few daily pills and/or a biweekly subcutaneous injection along with lab testing every two to three months rather than daily drops, periodic peri- or intraocular injections and likely multiple intraocular surgeries (with the attendant complications) over a treatment period that can extend five to 10 years.
Risks of Systemic Therapy
Unfortunately, the perception that systemic medications are “poisons” that consign a patient to future cancer risk or other complications may give some ophthalmologists or patients pause. Thankfully, good data exist. The Systemic Immunosuppressive Therapy for Eye disease (SITE) showed no increased overall or cancer-related mortality in uveitis patients treated with systemic immunomodulatory therapy.9
However, a small increased risk of skin cancer may exist, so counsel patients to wear sunscreen and protective clothing, pay attention to any new skin lesions, and undergo a periodic skin exam.10
When to Use Systemic Therapy
A pattern of frequent and significant disease recurrence disrupts a patient’s life and is often an indication for systemic therapy. Classic indicators are anatomic sequelae such as progressive synechiae and iris bombe or angle closure, steroid intraocular pressure (IOP) response, glaucomatous optic atrophy, cataract, vitreous opacity, uveitic macular edema, retinal capillary bed dropout, and macular
fibrosis associated with inflammatory choroidal neovascularization.
Other indications for
 |
| Figure 2. Visual fields of the 62-year-old woman with bird-shot retinochoroiditis in Figure 1 show extensive peripheral loss at the initial presentation. Visual field loss may be an impetus for systemic therapy, even in uveitis patients with good central vision and an edema-free macula. |
Diseases with a known poor prognosis (Behcet’s disease, bird-shot retinopathy and VKH) that inevitably smolder and relapse may be treated with systemic steroid-sparing therapy early on, even in the absence of a history of significant recurrence or sequelae.11 The earlier these severe patients begin systemic therapy, the better they do long term.
A Simplified Approach
The following systemic agents are indicated for infectious uveitis:
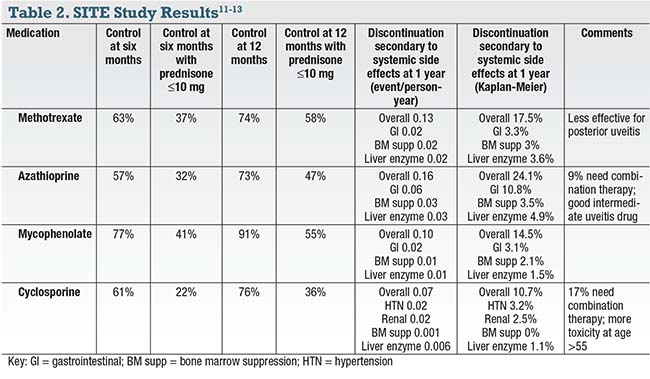
• Antimetabolites. The SITE study examined the use of azathioprine,12 methotrexate13 and mycophenolate (Table 2).14 One lesson from the SITE study is that control rates increase from six to 12 months, so you should emphasize patience and a long-term outlook in patient discussions.
The main side effects of antimetabolites are gastrointestinal (GI) upset, bone marrow suppression and liver enzyme abnormalities. GI upset will often improve after one to two weeks of therapy, but some patients will need to discontinue one agent after a couple weeks and try an alternative.
GI upset is idiosyncratic within the antimetabolite family. Some patients may develop symptoms with one agent but not another. Check complete blood count (CBC) and comprehensive metabolic profile (CMP) with a focus on liver enzymes at baseline and after two to three weeks of initial therapy. Thereafter, check labs every two to three months and three to four weeks after any dose increase.
Mycophenolate comes in 250-mg and 500-mg tablets. Typical dosing starts at 500 mg BID and ramps up to 1,000 mg BID in four to six weeks. Maximum dosing is 1,500 mg BID. Mycophenolate is popular among the antimetabolites because it may have a slightly lower incidence of side effects and thus is often the first choice for initial therapy.
Methotrexate comes in 2.5-mg or 5-mg tablets. Methotrexate may also be given as a subcutaneous injection. Regardless, methotrexate is dosed weekly, usually starting at 7.5 to 15 mg per week and then ramping up as needed. Maximum dose is 25 mg per week. Patients should take folate 1 mg the other six days of the week and either dramatically reduce or avoid alcohol intake. Methotrexate is less effective for posterior or panuveitis.
Azathioprine is available in 50-mg tablets. Dosing is based on body weight; 1-3 mg/kg/day, which usually translates to starting at 50 mg daily and increasing to 100 mg or 150 mg a day over the first one to three months. Avoid this medication in gout patients on allopurinol because it can magnify the bone marrow side effects. Azathioprine is a good intermediate uveitis drug and a decent posterior/panuveitis drug (Figure 3, page 26).
• T-cell inhibitors. Cyclosporine (Gengraf, AbbVie; Neoral and Sandimmune, Novartis) is a T-cell inhibitor with a slightly different side effect profile than the antimetabolites. I suggest avoiding Sandimmune, an older formulation. Tablets come as 100 mg or 25 mg. Dosing starts around 2 mg or 3 mg/kg daily and can go up to 5 mg/kg, typically
 |
Principal concerns with cyclosporine are renal toxicity, hypertension, liver enzyme abnormalities, low magnesium, elevated lipids and paresthesias. The side effect profile for cyclosporine is better than most physicians acknowledge. The drug tends to be well tolerated in patients younger than age 55 and can be used in older populations with close monitoring.
Patients should avoid heavy use of NSAIDS because they may magnify renal toxicity. Recommend frequent blood-pressure checks and ask patients to keep a log. Check blood count, blood chemistry with attention to serum creatinine and calculated creatinine clearance, and magnesium every one to two months initially and every two to three months thereafter.16 Patients should also have lipid profiles once or twice a year.
Tacrolimus (Prograf, Astellas) is a T-cell inhibitor with a side-effect profile and monitoring regimen similar to cyclosporine. While cyclosporine has a longer history for treatment of uveitis, tacrolimus may have a slightly lower incidence of kidney and blood-pressure issues.17 Conservatively, dosing ranges from 0.05 mg to 0.10 mg/kg daily. Tablets come in 1-mg size. Patients often started at 1 mg BID and may go up to 3 mg BID.
• Biologics. Infliximab (Remicade, Janssen), given as an intravenous infusion every four to six weeks, had been the prototypical anti-tumor necrosis factor (TNF) agent utilized for uveitis.18,19 Adalimumab (Humira, AbbVie), an anti-TNF agent that the patient typically self administers biweekly as a subcutaneous injection, has become a popular and effective option for uveitis, especially since the FDA granted it a labeled indication for noninfectious uveitis.20,21 Adalimumab is the only systemic non-corticosteroid agent FDA-approved for the treatment of noninfectious uveitis.
Anti-TNF agents may
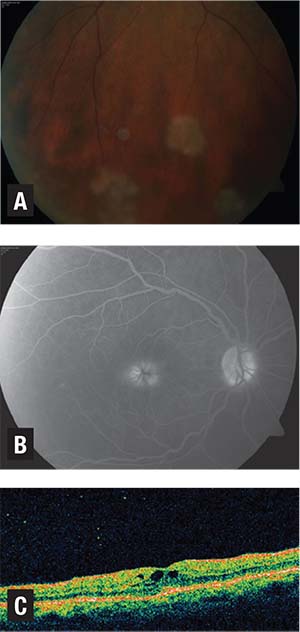 |
| Figure 3. Intermediate uveitis in a 63-year-old woman showing “taches de bougie,” or “candlewax drippings” (A). Fluorescein angiogram (B) shows late hyperfluoresence consistent with uveitic macular edema, and optical coherence tomography (C) shows prominent foveal cystoid spaces. Her pulmonary function studies were strongly suggestive of sarcoidosis, but she did not pursue lung biopsy. After local steroid therapy and four years of azathioprine therapy, her disease went into remission and she was successfully tapered off azathioprine. Courtesy Retina Center of Oklahoma |
Adalimumab is given as an 80-mg initial dose followed by 40 mg one week later and subsequently 40 mg biweekly. Patients should have negative QuantiFERON testing before they start therapy because the agent may reactivate latent tuberculosis infections. The retina subspecialist would do well to consult with a rheumatologist in prescribing and monitoring infliximab and adalimumab. These medications are mainstays in rheumatology.
Initial Drug Choice
An element of personal style shapes initial drug choice. In the last several years, mycophenolate has become increasingly popular as a first drug in all adult age groups. For intermediate uveitis, mycophenolate or azathioprine are good first-line choices. In young patients with significant posterior uveitis or panuveitis, cyclosporine either alone or in combination with mycophenolate or azathioprine is a good first choice. In older patients, the antimetabolite alone can be initiated. In children, methotrexate is often the first drug of choice.
Patient Counseling
Advise patients to be careful about sun exposure and to use sunscreen. They should also avoid live vaccines. While rare, any patient on immune suppression has a risk of John Cunningham virus-associated progressive multifocal leukoencephalopathy. Consult a neurologist if any new onset neurological symptoms develop.
Pregnancy-related Issues
Women of child-bearing age with uveitis that requires immunomodulatory therapy should be counseled on the need for proper contraception. Generally a hormonal method (eg, oral tablet or a depot injection) is inadequate, and patients typically are advised to use a second method concomitantly, usually a barrier method.
For retina specialists, communicating to the patient’s obstetrician-gynecologist or family doctor on the need for proper contraception associated with immunomodulatory therapy is typically sufficient. Verify with the patient on subsequent visits that she is compliant with the ob-gyn’s recommendations.
 |
When women with uveitis wish to become pregnant, systemic therapy should be tapered to end at least three months before attempting conception. Despite some discussion that agents like cyclosporine may be used in pregnancy, generally these patients should avoid immune suppression because pregnancy itself may have some immune-suppressive effects. Topical, injected or oral steroids can often manage flares during pregnancy.
Children with Uveitis
A significant body of literature supports the use of systemic immunomodulatory therapy in children with uveitis.22,23 Children present special medical issues and dosing considerations, so collaboration is important—either with a pediatric rheumatologist, a pediatrician familiar with systemic therapy or an adult rheumatologist comfortable managing children.
Children usually tolerate systemic therapy quite well, so such therapy should be initiated early in the disease course. Conversely, local steroid injections in children often quickly stimulate steroid-related cataract and ocular hypertension, prompting surgery that may provoke additional inflammation and anatomic complications, causing a downward clinical spiral. Local steroid injections have a role for treatment of uveitis in children, but they should be used judiciously and sparingly.
In the setting of macular edema, a three-to-six week course of oral steroids (dosed at 0.5 mg to 1 mg/kg daily) will often treat the macular edema in the short term and avoid the need for a steroid injection. Short-term oral steroids generally carry a lower risk of cataract and glaucoma than an injection. The use of oral steroids in such fashion allows time for systemic therapy to start to take effect.
In children with pars planitis, the pars plana membrane will involute slowly over one to three years. Look for less localized haze overlying the membrane, a more gray and less white coloration to the membrane, and decreasing thickness of the membrane. Vitreous cells often persist at the 1 to 2+ level in these patients, raising the question if the medication is having its intended effect.
Again,
 |
Efficacy and Dose Adjustment
Nothing is perfect; expect inflammatory breakthroughs during systemic therapy, most of which tend to be fairly mild and treatable with topical, injected or oral steroids. The vitreous need not be cell free. In my experience (other subspecialists may disagree), patients may have 1 to 2+ vitreous cell and be “controlled” and stable for the long term: the anterior chamber has ½+ or fewer cells; the vitreous cells, while up to 2+, are relatively fixed in the gel and pigmented; and the eye lacks vitreous haze, significant retinovascular leakage on fluorescein angiography (FA) or anatomic edema on OCT. Vision is typically good and the patient notes the occasional floater but overall good subjective visual function.
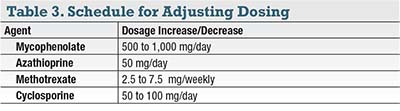
Use all available metrics as indicated for the particular disease—exam, OCT, angiogram with sweeps or widefield FA. For diseases such as bird-shot retinopathy, serpiginous choroidopathy, VKH and AZOOR, consider a visual field every 12 months to assess response to therapy. Goldmann fields are ideal but difficult to obtain; typically an automated 30-2 Sita-Standard or a similar test will do. If a patient is lagging in one finding—for example, persistent uveitic macular edema on OCT or recurrent leakage and expansion of inflammatory choroidal neovascularization refractory to anti-VEGF therapy—consider increasing the dose (Table 3).
Document the date and magnitude of all dose changes carefully in the chart. The SITE study clearly shows that immunomodulatory therapy takes six to 12 months to take full effect.9 When increasing the dose, look for a biological signal of some efficacy in approximately three months, but remember the full effect of a dose increase may take six months (even 12) to manifest. Be patient and utilize oral steroids and local steroid injections judiciously to bridge the time for a dose change to take effect. Consider combination therapy as appropriate.
Safety and Dose Adjustment
Most patients on systemic therapy should have a CBC and CMP every two to four months. With regards to antimetabolites, if a lab comes back with a mildly depressed hemoglobin or white blood cell count, or a mildly elevated liver transaminase, do not panic. First, repeat the lab to verify the abnormal value. If the patient is otherwise doing fine, therapy may continue. Otherwise, reduce the dose by a significant increment (Table 3, page 27) and subsequently repeat the labs three or four weeks later. If the patient has other nonocular complications or the abnormal labs persist or worsen, consult an internist or rheumatologist.
Patients prone to high blood pressure (older age, family history, overweight, high stress or untreated sleep apnea) may develop some blood pressure elevation on cyclosporine or tacrolimus, but a mild elevation does not rule out use of these medications. Consult the patient’s internist. Underlying factors predisposing a patient to high blood pressure can be addressed and anti-hypertensive medication can often be prescribed. Likewise, if a patient on cyclosporine or tacrolimus shows a small bump in serum creatinine, consider an incremental dose reduction with reassessment of the lab values one to two months later.
Immunomodulatory therapy may also continue in the setting of minor systemic infections. As a precaution, counsel patients to have a low threshold for seeing their primary-care doctor when ill, especially when symptoms persist. Primary-care doctors who know that a patient is on immunosuppression may be more prone to prescribe antibiotics for low-grade, community-acquired illnesses. The MUST trial reported a low rate of infections requiring treatment in both the implant and systemic therapy groups.7 The MUST investigators suggested these infections “were not associated with any lasting consequences” and hence overall mild.6,7
The Steroid-Sparing Effect
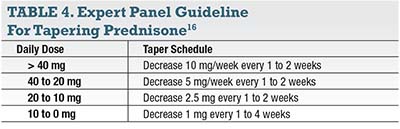
Many uveitis patients are on prednisone or need prednisone initially, so some suggestions for prednisone use are in order. Do not use the Medrol Dosepak (methylprednisolone, Pfizer); the duration is too short. Prescribe oral prednisone, usually 50 to 60 mg a day in adults (approximately 1 mg/kg/day). Ideally the patient should start a taper within four weeks, with a goal of 10 mg daily or less by three months. An expert panel developed guidelines for tapering prednisone (Table 4).15 Logically, the increment of taper decreases over time.
Chronic oral prednisone requires close attention to sugars, blood pressure and bone density issues. Supplements of calcium 1,000 to 1,500 mg a day and vitamin D3 800 to 1,000 iu a day are recommended.
• Going from 10 mg to zero. Most patients on adequate systemic therapy will taper to the low teens with regards to oral prednisone, but oftentimes that last 10 mg of prednisone can present a challenge. In difficult-to-wean patients, adopt a slow or “long-tail” taper for that last 10 mg. Decrease by 1 mg every one to two months. Taking a year to get to zero is acceptable in difficult patients. Establish a flare dose.
An example: if a patient on mycophenolate and cyclosporine makes it to 3 mg of daily oral prednisone and then has a mild breakthrough, put the prednisone back up to 10 mg, supplement as needed with local steroids, achieve quiescence again and then resume the taper a few weeks or months later. If the systemic agents are left at the same dose, come down to 4 or 5 mg a day (just above the flare dose) and then leave the patient at that level indefinitely, with the knowledge that such a low dose of chronic oral prednisone is fairly well tolerated. Not all patients will get to zero, but that’s acceptable.
Alternatively, at the time of the breakthrough, increase the dose of the steroid-sparing agents. Give that increased dose three to six months to take effect before attempting once again to dip below the “flare dose” and reach zero.
Role of Combination Therapy
As the SITE study demonstrated (Table 2), monotherapy works fairly well, but 20 to 40 percent of patients may need combination therapy, either because of progression or recurrences on monotherapy, a desire for steroid-tapering effect or disease severity. Combination therapy is well tolerated. Toxicity is relatively low, especially in young people, who have the most at stake.
The traditional combination is an antimetabolite (methotrexate, mycophenolate or azathioprine) plus a T-cell inhibitor (cyclosporine or tacrolimus). An increasingly popular combination is an antimetabolite combined with an anti-TNF agent, usually adalimumab. Rheumatologists are quite comfortable with this combination and can collaborate with you.
Monotherapy often cannot stop difficult diseases such as the vascular dropout of Behcet’s disease or the chorioretinal scarring of VKH, sympathetic ophthalmia and serpiginous choroidopathy. Initial combination therapy is indicated in these cases. If the patient does quite well in the first year of combination therapy, one agent can be carefully tapered.
Tapering
If the immunomodulatory regimen has achieved disease quiescence of two years or more, you can consider a taper. The first step is to verify that the patient wants to taper therapy. Not surprisingly, patients enjoy the stability of disease quiescence and may choose to continue a regimen rather than risk a flare up.
The main rule is to proceed slowly. Usually by this time, you’ve followed the patient for several years and know the individual’s disease pattern well.
In the case of combination therapy, taper the weaker agent in a multi-agent regimen first—but any toxicity issue takes precedence. Taper in small increments over six to 18 months. If you cannot achieve a full taper, do not view this as a failure; achieving a lower overall long-term dose is in and of itself a success. No shame exists in resuming a previously successful regimen if that’s what it takes to preserve long-term vision.
What to Tell Patients
Patients often struggle with a diagnosis of “uveitis.” After all, no one else they know has uveitis, and a lack of etiology after extensive medical testing can be frustrating. While the doctor should tell the patient the diagnosis is uveitis, it’s often helpful to say, “Uveitis is rheumatoid arthritis of the eye, and we use the same medicines that rheumatologists use in hundreds of thousands of people to treat arthritis.”
You may also say, “The immune system can be overactive or autoimmune in any part of the body, for reasons we don’t fully understand, and autoimmune eye disease is called uveitis.” These explanations allow uveitis patients to relate to the larger and better known group of patients with autoimmune and rheumatologic diseases and often alleviate anxiety.
And lastly for physicians, do not let a lack of etiology delay therapy. Do not hesitate to initiate systemic therapy in lieu of repeated steroid injections. Consider the analogy of transplantation. We’ve all been asked by a blind patient, “Doctor, is an eye transplant available?” If an ophthalmologist developed a technique of eye transplantation, that surgeon would win every award in our field and be hailed as a hero. No one would question the use of immunosuppressive drugs for “eye transplant patients”—but the use of these immunosuppressive drugs to preserve vision in uveitis patients has a 35-year track record. RS
REFERENCES
1. Thorne JE, Suhler E, Skup M, et al. Prevalence of noninfectious uveitis in the United States: A claims-based analysis. JAMA Ophthalmol. 2016;134:1237-1245.
2. Interpretive Handbook Test 34510: Syphilis IgG antibody with reflex, serum clinical information. Mayo Clinical Mayo Medical Laboratories. http://www.mayomedicallaboratories.com/interpretive-guide/?alpha=S&unit_code=34510 Accessed May 9, 2017.
3. Test Center: Syphilis antibody cascading reflex. Quest Diagnostics. http://www.questdiagnostics.com/testcenter/BUOrderInfo.action?tc=90349&labCode=DLO Accessed May 9, 2017
4. Nguyen QD, Merrill PT, Clark WL, et al. For the Sirolimus study Assessing double-masKed Uveitis tReAtment (SAKURA) Study Group. Intravitreal sirolimus for noninfectious uveitis: A Phase III Sirolimus Study Assessing Double-masKed Uveitis TReAtment (SAKURA). Ophthalmology. 2016;123:2413-2423.
5. Jaffe GJ, Lin P, Keenan RT, Ashton P, Skalak C, Stinnett SS. Injectable fluocinolone acetonide long-acting implant for noninfectious intermediate uveitis, posterior uveitis and panuveitis: Two-year results. Ophthalmology. 2016;123:1940-1948.
6. Multicenter Uveitis Steroid Treatment (MUST) Trial Research Group. Kempen JH, Altaweel MM, Drye LT, et al. Benefits of systemic anti-inflammatory therapy versus fluocinolone acetonide intraocular implant for intermediate uveitis, posterior uveitis, and panuveitis: Fifty-four-month results of the Multicenter Uveitis Steroid Treatment (MUST) Trial and Follow-up Study. Ophthalmology. 2015;122:1967-1975.
7. Multicenter Uveitis Steroid Treatment (MUST) Trial Follow-up Study Research Group. Quality of life and risks associated with systemic anti-inflammatory therapy versus fluocinolone acetonide intraocular implant for intermediate uveitis, posterior uveitis, or panuveitis: Fifty-four-month results of the Multicenter Uveitis Steroid Treatment Trial and Follow-up Study. Ophthalmology. 2015;122:1976-1986.
8. Writing committee for the Multicenter Uveitis Steroid Treatment (MUST) Trial and Follow-up Study Research Group. Association between long-lasting intravitreous fluocinolone acetonide implant vs systemic anti-inflammatory therapy and visual acuity at 7 years among patients with intermediate, posterio, or panuveitis. JAMA. 2017;317:1993-2005.
9. Kempen, J.H., Daniel, E., Dunn, J.P. et al, Overall and cancer related mortality among patients with ocular inflammation treated with immunosuppressive drugs: retrospective cohort study. BMJ.2009;339:b2480
10. Yates WB, Vajdic CM, Na R, McCluskey PJ, Wakefield D. Malignancy risk in patients with inflammatory eye disease treated with systemic immunosuppressive therapy. Ophthalmology. 2015;122:265-273.
11. Bykhovskaya I, Thorne JE, Kempen JH, Dunn JP, Jabs DA. Vogt-Koyanagi-Harada disease: clinical outcomes. Am J Ophthalmol. 2005 ;140:674-678
12. Pasadhika S, Kempen JH, Newcomb CW, et al. Azathioprine for ocular inflammatory diseases. Am J Ophthalmol 2009;148:500-509.e2.
13. Gangaputra S, Newcomb CW, Liesegang TL, et al. For the Systemic Immunosuppressive Therapy for Eye Diseases Cohort Study. Methotrexate for ocular inflammatory diseases. Ophthalmology. 2009;116:2188-9218.e1.
14. Daniel E, Thorne JE, Newcomb CW, et al. Mycophenolate mofetil for ocular inflammation. Am J Ophthalmol. 2010; 149:423-432.
15. Kaçmaz RO, Kempen JH, Newcomb C, et al. Cyclosporine for ocular inflammatory diseases. Ophthalmology. 2010;117:576-584.
16. Jabs DA, Rosenbaum JT, Foster CS, et al. Guidelines for the use of immunosuppressive drugs in patients with ocular inflammatory disorders: recommendations of an expert panel. Am J Ophthalmol. 2000;130:492-513.
17. Murphy CC, Greiner K, Plskova J, et al. Cyclosporine vs tacrolimus therapy for posterior and intermediate uveitis. Arch Ophthalmol. 2005;123:634-41.
18. Kruh JN, Yang P, Suelves AM, Foster CS. Infliximab for the treatment of refractory noninfectious Uveitis: a study of 88 patients with long-term follow-up. Ophthalmology. 2014;121:358-364.
19. Takeuchi M, Kezuka T, Sugita S, et al. Evaluation of the long-term efficacy and safety of infliximab treatment for uveitis in Behçet’s disease: a multicenter study. Ophthalmology. 2014;121:1877-1884.
20. Levy-Clarke G, Jabs DA, Read RW, Rosenbaum JT, Vitale A, Van Gelder RN. Expert panel recommendations for the use of anti-tumor necrosis factor biologic agents in patients with ocular inflammatory disorders. Ophthalmology. 2014;121:785-796.e3.
21. LaMattina KC, Goldstein DA. Adalimumab for the treatment of uveitis. Expert Rev Clin Immunol. 2017 Mar;13(3):181-188.
22. Gregory AC 2nd, Kempen JH, Daniel E, et al. For the Systemic Immunosuppressive Therapy for Eye Diseases Cohort Study Research Group. Risk factors for loss of visual acuity among patients with uveitis associated with juvenile idiopathic arthritis: the Systemic Immunosuppressive Therapy for Eye Diseases Study. Ophthalmology. 2013;120:186-92.
23. Hersh AO, Cope S, Bohnsack JF, Shakoor A, Vitale AT. Use of immunosuppressive medications for treatment of pediatric intermediate uveitis. Ocul Immunol Inflamm. 2016 Dec 14:1-9.
24. Malalis JF, Bhat P, Shapiro M, Goldstein DA. Retinoschisis in pars planitis. Ocul Immunol Inflamm. 2016;22:1-5.



